一对称性色素异常症家系ADAR基因突变分析及产前诊断
2021-01-12秦艳芳朱留洋王聪慧孔祥东
秦艳芳, 朱留洋, 王聪慧, 孔祥东
(1.濮阳市油田总医院,河南 濮阳 457001;2.郑州大学第一附属医院,河南 郑州 450052)
遗传性对称性色素异常症(dyschromatosis symetrica hereditaria,DSH),也称肢端色素沉着症,在各个种族中可见,但多见于亚洲人,其中中国和日本报道较多[1]。该病主要临床表现为肢端伸侧出现对称性色素减退斑和色素沉着斑,尤其是手、足背部明显,有时面部出现雀斑样皮损,可不伴色素减退斑,常在婴儿期或儿童早期出现,青春期表现较为明显,发病进程比较缓慢,病程伴随终生[2-3]。本病一般无自觉症状,日晒可加重其症状。致病基因为ADAR基因。其组织病理学无典型改变,活检发现色素减退斑处黑素缺失时,电镜下可观察到黑素细胞状态异常;活检见色素沉着斑处基底层黑素增多时,电镜下可见黑素细胞代谢活性增高、细胞增多,黑素细胞拥有更多的树突和大量的Ⅳ期黑素小体[4]。
通过对不同家系及散在发病者的研究,迄今已发现二百多种不同的突变,包括错义突变、无义突变、移码突变、剪接区突变和复杂的插入/缺失突变等[5]。导致发病的主要是错义突变,发生在第9~15外显子的突变达60%以上[6]。本研究对一中国汉族DSH家系行ADAR基因突变检测,分析其致病性及作出产前诊断。
1 资料与方法
1.1 临床资料
先证者,男,6岁,颈部、双侧关节、双手背和足背处色素异常5年余。患者7个月时颈部、双侧关节、双手背和足背处出现散在色素减退斑和沉着斑,针尖及粟粒大小, 随着年龄增长皮疹面积逐渐增大,并蔓延至双前臂伸侧、臀部、面部,尤其手部为重。表现为手足背、颈、臀部不规则色素沉着或减退斑,颜色深浅不一、部分融合成网状(图1),日晒后皮损可加重。黏膜、指(趾)甲、牙齿和毛发均正常。先证者祖母,自幼发病,未在意,生育后于外院行皮损活检诊断为DSH,现已去世,资料丢失。先证者父亲,1岁时发病,未予诊治。先证者母亲,30岁,再次妊娠,孕18周,拟通过产前诊断检测胎儿是否患DSH,就诊于郑州大学第一附属医院遗传与产前诊断中心。本研究经郑州大学第一附属医院医学伦理委员会批准(批件号:KS-2018-KY-36),患者及家属同意并签署知情同意书。
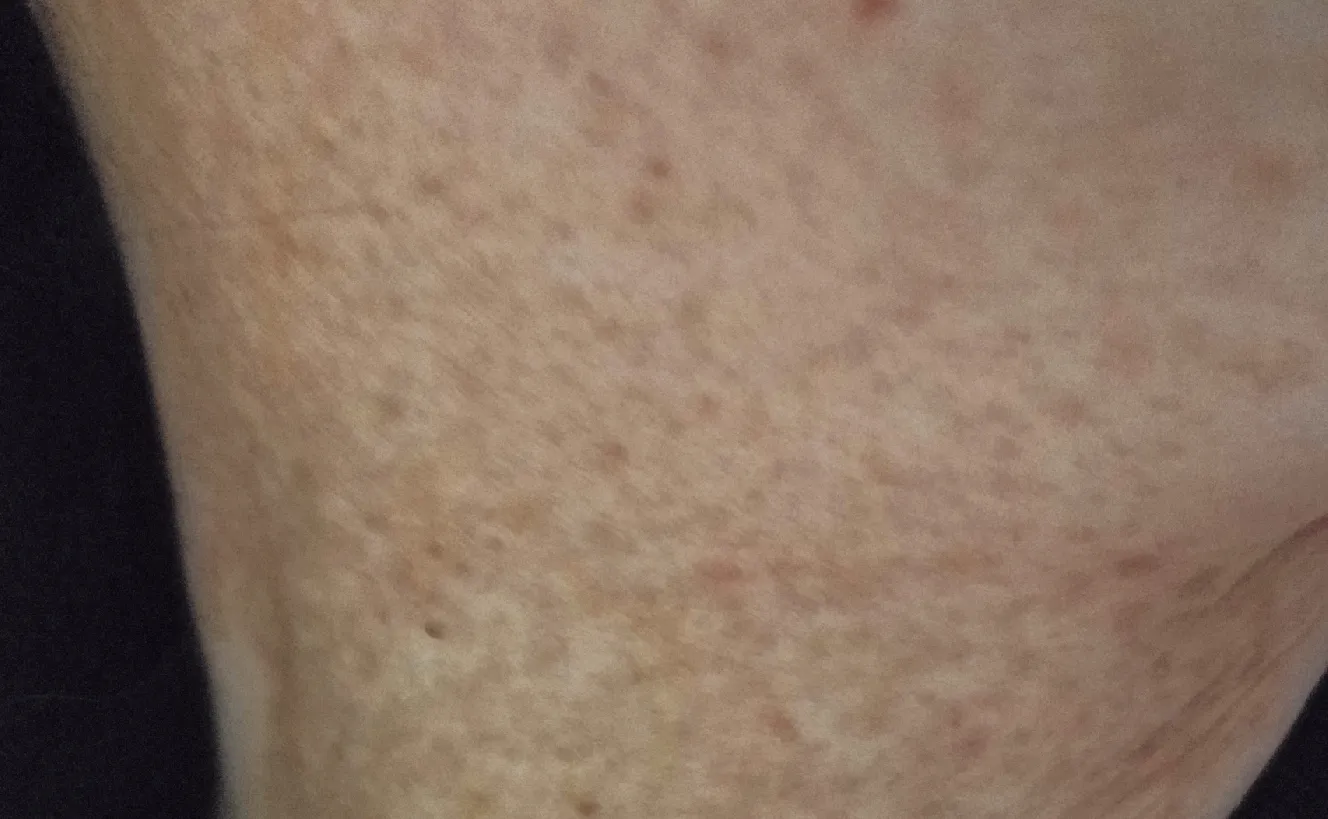
图1 先证者颈部右侧临床图片
1.2 方法
1.2.1 基因组DNA提取 采集其家系10个成员(先证者及其父亲、母亲、姑姑、姑父、表姐、叔叔、婶婶、堂姐、堂哥)静脉血各2 mL,采集先证者母亲羊水20 mL,采用美国Promega公司DNA提取试剂盒提取基因组DNA。
1.2.2 高通量测序及Sanger测序验证 利用Ion Personal Genome Machi-ne(PGMTM)测序仪进行高通量测序。目标捕获区平均测序深度为126 X,测序覆盖度为98.21%,测序深度大于100 X的靶向读长达100%,根据ACMG的遗传变异分析指南对所有检测到的变异进行筛选,设计引物并对突变位点进行Sanger测序,分析结果。
1.2.3 引物设计 根据ADAR基因第二外显子编码区和侧翼序列设计引物,均由上海生工生物工程有限公司合成。上游引物:5′-CGGAAGACAGAAACTCCAC-3′,下游引物:5′-CTGCTC ACAAATCAGCCAAG-3′。
1.2.4 PCR扩增 扩增条件:94 ℃预变性5 min;94 ℃变性45 s,58 ℃退火1 min,72 ℃延伸1 min,共33个循环;72 ℃延伸10 min。扩增结束后,PCR产物经琼脂糖凝胶电泳鉴定、纯化后直接在3130XL一代测序仪(Life公司)上测序,所有测序结果与dbsnp.1000Genomes Project等数据库相关数据进行分析比对。
2 结果
2.1 家系调查
经调查,该家系3代有3例发病(图2),男女比例为2 ∶1,结合该家系遗传史、典型临床表现及单基因病种检测,确诊为常染色体显性遗传DSH。

图2 家系谱
2.2 基因变异定性分析及家系胎儿产前诊断
结果显示,先证者、先证者父亲、先证者母亲羊水中均携带ADAR 基因c.613C>T(p.Q205X)位点杂合变异,而家系中其他人员未见该改变(图3)。该突变在dbSNP数据库、1000Genomes Project 数据库中均未找到。使用polyphen2(http://genetics.bwh. Harvard. edu/pph2/),SIFT(http:// sift. jcvi.org/)和Mutation taster( http: // www. Mutatio -ntaster.org/)等生物信息软件对该突变进行致病性预测,结果表明,ADAR 基因c.613C>T(p.Q205X)位点杂合变异是该家系的致病突变而不是单核苷酸多态性。

图3 基因测序结果Fig.3 Gene sequencing results.
3 讨论
随着DSRAD/ADAR基因确定为DSH的致病基因,对于不同家系致病基因突变位点的检测已成为研究DSH的一个热点。ADAR基因总长30 kb,有15个外显子,可编码1 226个氨基酸[7]。ADAR也称双链RNA特异性腺苷脱氨酶,该酶选择性作用于mRNA前体,将相应位点上腺嘌呤核苷(A)脱氨基转换成次黄嘌呤核苷(I)[8]。含有2个腺苷脱氨基酶Zalpha区域、3个ds-RNA结合区域和1个脱氨基区域,分别对应于外显子2、外显子2-7和外显子9-15[9-10]。该基因不但将mRNA前体特异位点的A编辑为I,还可编辑双链RNA中约40%~50%的An41[11]。彭琛等[7]对一DSH 并发银屑病家族进行基因检测,发现先证者及家系中其他患者ADAR1 基因均存在第8外显子突变,家系中非患病者及无家系关系的对照组均未发现该突变。突变 c.2745_2746del CT(p.D582X),即在第 8 外显子中第 2 745~2 746 位碱基C及碱基T缺失,由于框移突变造成第582位密码子由TCT变成TGA,终止密码子提前出现,导致其后12个氨基酸缺失,酶的催化功能受损,引起DSH的发生。
DSH是一种常染色体显性遗传病,但也存在散发病例[12]。曾荣等[5]提取一家系中2例DSH患者外周血DNA,以基因组DNA为模板扩增出PCR产物,并纯化后测序,将患者ADAR1基因全部编码区测序结果比对数据库的序列,结果显示,2例患者ADAR1基因错义突变(c.662C>T),引起ADAR1基因中编码的色氨酸(CGG)变成精氨酸(TGG),而家系中未患病及50位与本家系无关的正常人未见该改变。本研究中,该家系3代12人中3例发病,孕18周的胎儿经检测为确诊患者,每代均有发病,遗传方式符合常染色体显性遗传,无论胎儿为哪种性别,其再发风险高达50%。
本研究将PCR产物测序结果与polyphen2、SIFT和 Mutation taster中相对应的正常人ADAR基因序列进行比对,检测到该家系中患者出现杂合突变,ADAR基因的第2号外显子613位碱基胞嘧啶(C)突变成胸腺嘧啶(T),造成编码氨基酸的基因发生无义突变,最终导致第205位氨基酸形成终止密码子, 即p.Q205X,使ADAR基因所编码的蛋白提前终止。在表达过程中截短蛋白的产生导致其后大片氨基酸缺失,或可能引起无义介导的mRNA衰减,减少ADAR蛋白的表达,造成单倍剂量不足,促使酶蛋白的催化功能异常,最终导致黑素母细胞功能异常,从而产生DSH相应的临床症状。先证者及先证者父亲根据临床表现诊断DSH,而临床表型正常的先证者母亲未发现突变,表明发现的无义突变(c.613C>T)是该患者的致病突变而不是单核苷酸多态性。胎儿ADAR基因检测显示与先证者及先证者父亲基因检测结果相同,皆出现c.613C>T(p.Q205X)位点无义突变,从而亦确诊为DSH。
经查阅国内外文献发现,本研究报告的c.613C>T(p.Q205X)为未曾报道过的无义突变,该突变扩大了DSH的基因谱。通过基因测序获得的大量突变数据,可为DSH的基因型与表型关联、致病机制及治疗等进一步研究提供帮助。DSH因病变部位暴露的皮肤影响美观,常给患者带来心理负担。因此,防治关键是检测其致病基因,找出致病基因的突变位点进行产前筛查,同时作出产前诊断,预防下一代患者的发生。
产前诊断技术是指利用超声检查、母体血清学筛查、无创产前基因检测、胎儿染色体核型分析、荧光原位杂交、染色体芯片分析及二代测序等技术对胚胎或胎儿的发育情况及是否患病等方面进行检测诊断[13]。利用产前诊断技术可尽早发现缺陷胎儿,并判断其能否继续妊娠,从而降低出生缺陷率,减轻家庭和社会的经济和精力负担、提高生活质量等。本研究中,先证者父母再次怀孕时,为避免生育遗传性对称性色素异常患者而进行产前诊断。先证者长大成人婚育前建议进行遗传咨询。
致谢:感谢郑州大学第一附属医院老师在实验过程中的帮助及指导。
