新生儿异常Hb Q的家系分析*
2022-06-22葛艳芬刘均如黄革李广华周茂华林婷冼璐桦
葛艳芬 刘均如 黄革 李广华 周茂华 林婷 冼璐桦
血红蛋白病是因血红蛋白质和量的缺陷而导致的一类常染色体隐性遗传性疾病,其可分为两类:一类是异常血红蛋白病,是血红蛋白发生结构上的异常而引发溶血症,通常是由于α-或β-珠蛋白链单个氨基酸替换产生异常血红蛋白,即功能和质量改变;另一类是地中海贫血(简称地贫),是由某种珠蛋白合成受抑制引起的贫血,即数量缺失[1]。常表现为溶血性贫血,主要类型有α-、β-和δ-地贫等。据报道,广州地区地贫基因的携带率为11.25%,其中α-地贫基因携带率为7.76%,β-地贫基因携带率为3.02%,α-地贫复合β-地贫基因携带率为0.49%,广东省异常血红蛋白的发病率0.368%[2-3]。在我国南方地区,较常见的异常血红蛋白病主要以Hb Q、Hb E及Hb New York为主[4]。其中Hb Q是一种少见的α链变异的异常血红蛋白,本研究分析了5例新生儿Hb Q家系,现报道如下。
资料与方法
1 一般资料 先证者1-5均为1 d的新生儿,其中先证者1-2为男,3-5为女;先证者1、3、4和5及其父母籍贯分别为辽宁、广东、江西、甘肃,均为其母生产后正常送检新生儿血样本,经血红蛋白电泳发现其S区含有异常血红蛋白区带;先证者2及其父母籍贯广东。其父母在产检时发现为α地贫携带者,被告知胎儿可能患有Hb H病。家系成员的受检样品均行血常规检测,Heinz小体试验,Hb电泳与α、β地中海贫血基因检测。
2 仪器与试剂 Capillarys 2 flex- piercing全自动毛细管电泳仪及其配套试剂购自法国Sebia公司。
Beckman LH 750、Sysmex XS-800i全自动血液分析仪及其配套试剂分别由相应公司提供。
3 毛细管电泳分析 按照Capillarys 2 flex- piercing全自动毛细管电泳仪的操作要求完成质控。将ACD管抗凝血样品3 000 r/min离心2 min,将血浆和血细胞配成1∶1,摇匀后放入全自动毛细管电泳仪,进行血红蛋白电泳。在9.8 kV电压、pH 9.4的碱性缓冲液条件下,于石英毛细管内进行血红蛋白电泳,用415 nm波长检测各种血红蛋白的百分比,进而对血红蛋白定量。
4 血常规检测 按照Beckman LH 750、Sysmex XS-800i全自动血液分析仪操作要求完成定标与质控,将EDTA-K2管抗凝血直接上机检测。
5 地中海贫血检测 地中海贫血基因:采用深圳益生堂公司的缺失型α-地中海贫血基因诊断试剂盒,运用 gap-PCR技术,以基因组DNA为模板,应用单管四重PCR/琼脂糖凝胶电泳技术,检测--SEA、-α3.7、-α4.2三种常见的缺失型α-地贫基因型,同时选用反向斑点杂交(RDB)法检测3种常见的非缺失型α-地中海贫血基因型: -αCS、-αQS、-αWS。β地中海贫血基因:采用反向斑点杂交(RDB)法检测17种常见的突变及其正常序列: CDs41-42M、IVS2-654M、-28M、CD17M、CDs71-72M、βEM、CDs27-28M、-29M、CDs14-15M、CD43M、IVS1-1M、IVS1-5M、IntM、-30M、CD31M、-32M、CAP。
结 果
1 血液学与地贫基因分析结果 先证者1 MCV:90.4 fL,MCH:29.8 pg,其父MCV:76.1 fL,MCH:25.6 pg,先证者1及其父α基因型均为-α4.2/αα;先证者2 MCV:75.1 fL,MCH:22.6 pg,其父MCV:66.3 fL,MCH:20.5 pg,先证者2及其父、母α基因型分别为--SEA /-α4.2、--SEA /αα、-α4.2/αα。先证者3 MCV:93.0 fL,MCH:32.5 pg,其母MCV:80.3 fL,MCH:26.1 pg,两者α基因型均为-α4.2/αα。先证者4 MCV:92.9 fL,MCH:30.3 pg,其母MCV:76.2 fL,MCH:25.1 pg,两者α基因型均为-α4.2/αα。先证者5 MCV:89.4 fL,MCH:29.9 pg,其母MCV:78.9 fL,MCH:26.8 pg,两者α基因型均为-α4.2/αα。见表1。
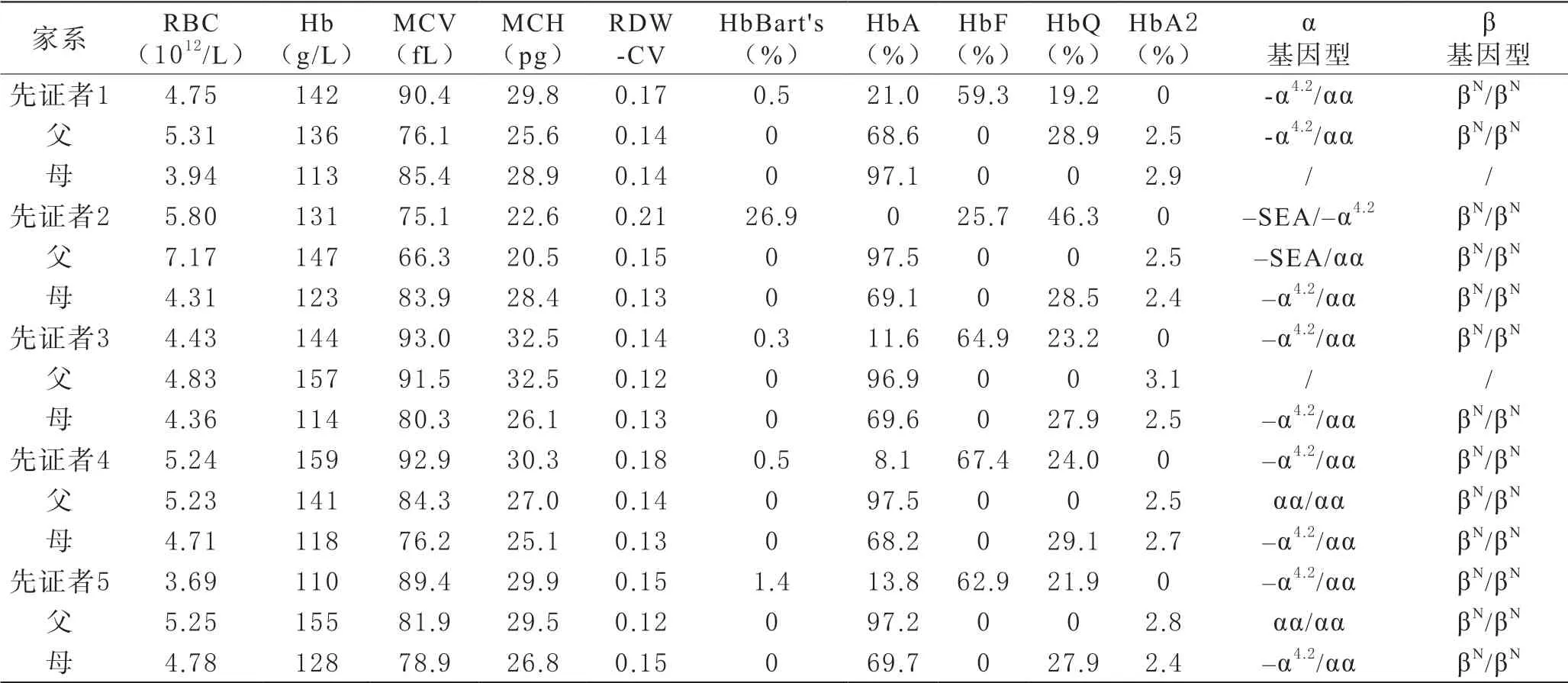
表1 先证者及其父母血液学及基因检测结果
2 Hb电泳分析图谱 先证者1的Hb电泳在S区含有19.2%的区带,Hb Bart's:0.5%,Hb A:21.0%,Hb F:59.3%。其父为异常Hb Q1含量:28.9%,Hb A:68.6%,Hb A2:1.8%,Hb Q2:0.7%;先证者2的Hb电泳在S区含有46.3%的区带,Hb Bart's:26.9%,Hb F:25.7%,未见Hb A;其母为异常Hb Q1含量:28.5%,Hb A:69.1%,Hb A2:1.7%,Hb Q2:0.7%;其父Hb A:97.5%,Hb A2:2.5%。先证者3的Hb电泳在S区含有23.2%的区带,Hb Bart's:0.3%,Hb A:11.6%,Hb F:64.9%;其母为异常Hb Q1含量:27.9%,Hb A:69.6%,Hb A2:1.8%,Hb Q2:0.7%。先证者4的Hb电泳在S区含有24.0%的区带,Hb Bart's:0.5%,Hb A:8.1%,Hb F:67.4%;其母为异常Hb Q1含量:29.1%,Hb A:68.2%,Hb A2:1.9%,Hb Q2:0.8%。先证者5的Hb电泳在S区含有21.9%的区带,Hb Bart's:1.4%,Hb A:13.8%,Hb F:62.9%;其母为异常Hb Q1含量:27.9%,Hb A:69.7%,Hb A2:1.7%,Hb Q2:0.7%。见图1-5。3 Heinz小体试验结果 先证者2的Heinz小体试验阳性,阳性率为0.5%,其父亦为阳性,因量少,未计算阳性率。余受检者均未检出。

图1 先证者1、父、母Hb电泳图谱
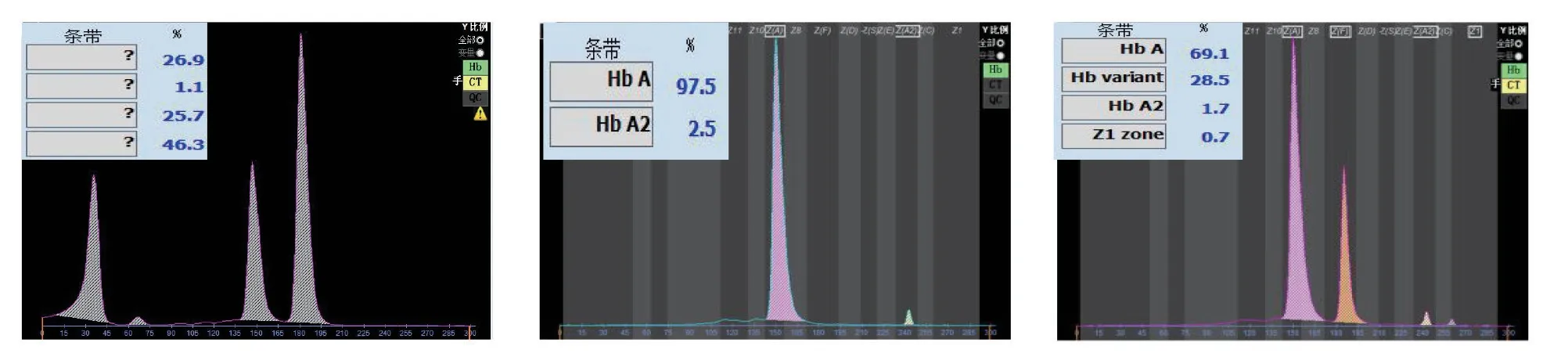
图2 先证者2、父、母Hb电泳图谱
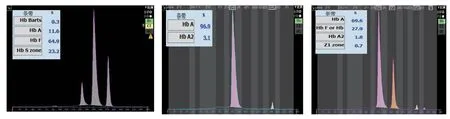
图3 先证者3、父、母Hb电泳图谱

图4 先证者4、父、母Hb电泳图谱
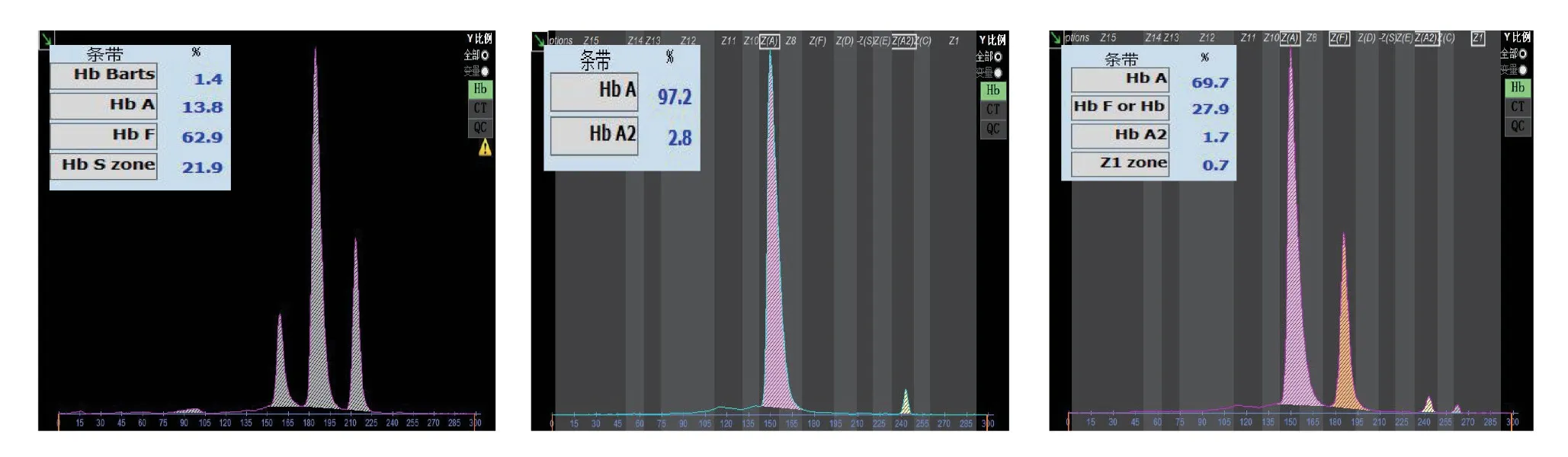
图5 先证者5、父、母Hb电泳图谱
讨 论
异常Hb Q是我国少见的一种异常血红蛋白变异体,属于α链变异,其α1链N端第74位天冬氨酸被组氨酸取代,最早在新加坡一个华人家庭中发现[5],中国最先由曾溢滔等[6]发现。与其他异常血红蛋白不同的是,其常与-α4.2/αα缺失型的α地中海贫血连锁携带[7-8]。绝大部分异常血红蛋白杂合子临床表型不明显或者无临床表型,血常规无法检出,一般都是通过血红蛋白电泳检出异常带。目前较好的实验室检测方法为全自动毛细管电泳,全自动毛细管电泳不仅能筛查出常见地贫患者产生的异常血红蛋白条带,而且对罕见的地贫基因携带者产生的异常血红蛋白条带敏感,能有效地分离和准确定量Hb Q、Hb E、Hb J、Hb D、Hb New York、Hb CS、Hb H和Hb Bart's等异常血红蛋白[9]。值得注意的是,目前实验室所使用的地贫基因检测试剂盒无法检出Hb Q,而只能检测出其连锁的-α4.2缺失型。若不进行血红蛋白电泳,极易将患者误判为只有-α4.2缺失杂合子,导致异常血红蛋白的漏检。
本研究先证者1 Hb电泳发现在S区含有19.2%的区带,Hb Bart's:0.5%,Hb A:21.0%,Hb F:59.3%。其父为异常Hb Q1:28.9%,Hb A:68.6%,Hb A2:1.8%,Hb Q2:0.7%。先证者2其母为异常Hb Q1含量:28.5%,Hb A:69.1%,Hb A2:1.7%,Hb Q2:0.7%。先证者3 Hb电泳发现在S区含有23.2%的区带,Hb Bart's:0.3%,Hb A:11.6%,Hb F:64.9%。其母为异常Hb Q1:27.9%,Hb A:69.6%,Hb A2:1.8%,Hb Q2:0.7%。先证者4 Hb电泳发现在S区含有24.0%的区带,Hb Bart's:0.5%,Hb A:8.1%,Hb F:67.4%。其母为异常Hb Q1:29.1%,Hb A:68.2%,Hb A2:1.9%,Hb Q2:0.8%。先证者5 Hb电泳发现在S区含有21.9%的区带,Hb Bart's:1.4%,Hb A:13.8%,Hb F:62.9%。其母为异常Hb Q1:27.9%,Hb A:69.7%,Hb A2:1.7%,Hb Q2:0.7%。先证者1、3、4和5有Hb Bart's区带且量较少,推测其为静止型的α地贫。根据连锁遗传规律、地贫基因和家系分析,初步判断先证者1及其父亲、先证者2其母、先证者3-5及其母为Hb Q合并静止型的α4.2地贫。Hb Q杂合子有部分红细胞参数正常,当合并地贫时,Hb Q杂合子可表现出红细胞平均体积和平均血红蛋白含量降低或不同程度贫血,与轻型地贫血液学参数类似[10-11]。先证者1、2、3、4和5的MCV值分别为90.4、75.1、93.0、92.9及89.4 fL(新生儿正常参考范围一般>100 fL),先证者1其父,先证者3-5其母MCV值分别为76.1、80.3、76.2及78.9 fL,MCH为25.6、26.1、25.1及26.8 pg,均提示减低,与文献报道相一致[12-13]。
该基因携带者与--SEA/αα地中海贫血基因携带者结合时,25%发生Hb Q-H病,先证者2为Hb Q-H病患者,由于Hb Q稳定且携氧能力不低,不会加重患者的临床症状。因此,Hb Q-H病与Hb H病在症状上相似,另也有文献报道其可能会加重地贫患者的贫血程度,但Hb Q-H合并β地贫时临床症状可减轻[14-15]。Hb Q-H病是有三个α基因缺失,剩下一个α直接变成αQ,因此Hb电泳图是没有Hb A。先证者2就属于这种情况。新生儿的Hb H病,Hb Bart's量一般大于15%,先证者2 Hb Bart's量达到26.9%,Heinz小体0.5%,可以初步推断是Hb H病,且基因结果为--SEA /-α4.2,证实其是Hb H病。结合Hb电泳S区含有26.9%区带,分析其为Hb Q-H病。
同样异常Hb Q,先证者1-5的Hb电泳位置在S区,其父或母在F区,可能新生儿主要以Hb F为主,而成人以Hb A为主,导致电泳位置有所不同。由此可见新生儿异常Hb Q很容易误判为Hb S,尤其对于初学者应引起关注,需根据家系分析其异常Hb。异常Hb可通过DNA测序技术确诊,而常规地贫基因无法检测。但因条件所限,尤其基层医院,毛细管电泳是可行的检测方法,其可对人群异常血红蛋白进行筛查,并根据图谱推测疑似Hb属于α链或β变异,为基因测序方法顺利进行提供依据。
综上所述,广东地区地贫高发,将导致Hb Q复合地贫可能性增大,因此要求医务人员在日常工作中务必重视Hb Q患者及其他异常Hb携带者,做好患者遗传咨询,达到优生优育目的。
利益冲突所有作者均声明不存在利益冲突
