PCDH15突变导致遗传性耳聋的研究进展
2022-02-28施韬关静王秋菊
施韬关静王秋菊*
1浙江中医药大学(杭州 310053)
2中国人民解放军总医院耳鼻咽喉头颈外科医学部,国家耳鼻咽喉疾病临床医学研究中心,解放军耳鼻咽喉研究所,聋病教育部重点实验室;聋病防治北京市重点实验室(北京 100853)
人体许多细胞可感受机械性刺激并转换成电信号。这种功能的实现取决于机械门控通道,它对人的听觉、保持平衡,感知环境危机等极其重要。比如耳蜗毛细胞的静纤毛受到剪切力的刺激时会发生弯曲,进而打开相关离子通道,产生短暂的感受器电位,并随着听神经向中枢传递[1,2]。在耳蜗毛细胞机械传导机制中,静纤毛间的顶连接张力的改变可以改变机械门控通道打开的概率和程度[3]。
PCDH15编码的原钙粘蛋白15(protocadherin 15,PCDH15)是顶连接的组成部分,PCDH15是钙依赖性细胞间粘附分子,在高级脊椎动物中,原钙粘蛋白与神经回路形成和突触形成等多种功能有关[4]。PCDH15变异将会导致Usher综合征1F型(USH1F)和常染色体隐性遗传性耳聋23型(DF‐NB23)。USH1是一种以进行性视网膜色素变性(retinitis pigmentosa,RP)和先天性感音神经性听力损失为特征的常染色体隐性疾病[5,6],伴有先天性前庭功能障碍,运动发育迟缓、难以保持平衡[7,8]。DFNB23患者仅表现为先天性双耳重-极重度耳聋[4,9]。本文旨在将PCDH15突变导致的DFNB23和USH1F研究进展进行综述,分析临床表现与基因型的关联以及它的致聋机制。
1 PCDH15的克隆、定位与表达
1997年,Wayne等发现Usher综合征新的基因座——USH1F,并将其定位于染色体10q21-22,标记D10S199-D10S596之间[10]。2001年,Alagramam等在Ames waltzer小鼠中发现Pcdh15突变,该基因突变导致小鼠静纤毛在出生后第10天出现异常以及内耳神经上皮细胞变性,最终出现耳聋及前庭功能障碍的表型[11]。Alagramam等使用小鼠Pcdh15 cDNA探针筛选人类基因组P1人工染色体文库,鉴定出三个人类同源基因PCDH15的克隆,并定位于染色体10q11.2-q21[5,11]。人和小鼠在10号染色体之间有高度的保守一致性,Pcdh15和PCDH15在胞外重复(extracellular cadherin,EC)结构域中的核苷酸水平上同源性近似85%,氨基酸序列的同源性近似94%[12]。2001年,Ahmed将导致USH1F的PCDH15定位于染色体10q21.1[13]。
Ahmed等通过Western blot揭示了Pcdh15蛋白在小鼠视网膜、耳蜗毛细胞和前庭毛细胞的表达,并且在不同的耳蜗毛细胞之间,以及同一毛细胞上不同长度的静纤毛中,Pcdh15蛋白的免疫反应的表达量是不同的[4]。对猫头鹰、小鼠视网膜细胞RNA测序,发现pcdh15在感光细胞中起重要作用[14,15]。Ahmed等发现在人眼的感光细胞中存在PCDH15蛋白,这表明USH1F患者可能在色觉方面存在重大缺陷,并且可能出现中心视力不适,夜盲症和进行性周围性视觉丧失等视觉障碍[4]。
2 PCDH15蛋白的结构与功能域
PCDH15基因位于10q21.1,含39个外显子,其编码的PCDH15蛋白属于钙粘蛋白的一个亚家族,包含11个EC结构域,1个跨膜结构域和细胞质结构域[2,7,16]。与经典钙粘蛋白不同的是,原钙粘蛋白的每一个EC结构域均由一个或多个外显子编码而成[9]。钙粘蛋白 23(cadherin-like 23,CDH23)和PCDH15分别形成同源二聚体,组成顶连接的上和下部,并通过N末端发生反式相互作用,该作用是Ca2+依赖性的。近期有研究提出了PCDH15与CDH23反式和顺式同源二聚体模型,并预测PCDH15具有刚性或柔性机械门控的特征[17]。
PCDH15最初鉴定出最长的转录本有33个外显子,编码包含1955个氨基酸的蛋白质,随后,Ahmed等鉴定出6个额外的外显子[7,13,18]。根据转录剪接的不同,PCDH15蛋白具有3种细胞质结构域的同工型蛋白[3,7]。这些同工型在毛细胞内表达的时间和位置均不同,说明它们在其中的作用是不同的[9]。在小鼠不成熟的听觉毛细胞中,3种Pcdh15同工型可以互相转换,以形成顶连接,但在小鼠成熟的听觉毛细胞中,Pcdh15-CD2是必不可少的[3]。
3 PCDH15突变与遗传性耳聋
PCDH15保守序列发生错义突变时,可能改变蛋白EC结构域的空间形态,扰乱PCDH15和CDH23之间的相互作用,从而破坏顶连接的完整性,静纤毛之间的相互作用减弱,机械门控通道出现异常,导致耳聋的发生[4,19]。目前国内外对DFNB23报道仅局限于少量家族报道(表1)。PCDH15基因的错义编码,影响耳蜗静纤毛间顶连接的正常功能,但PCDH15蛋白在前庭、眼部等处并未丧失全部功能,最终导致患者仅出现耳聋表型。2003年,Ahmed等首次报道在两个DFNB23家系中发现PCDH15的两种致病突变[4]。

表1 DFNB23家系中PCDH15突变Table 1 PCDH15 mutation in DFNB23 families
Usher综合征根据听力损失的发病年龄和严重程度,以及前庭功能是否异常,在临床上分为三个亚型,Usher综合征I型(USH1)是最严重的一种亚型,USH1型患者多是双耳先天性平坦型极重度听力损失、先天性前庭障碍以及进行性RP。USH1是一种基因异质性遗传疾病,至少11个基因位点与USH1有关,目前已经确定9个基因在染色体上的位置,其中PCDH15基因突变导致的USH1F是Usher综合征I型中第三常见的原因,在白种人当中约为9%-11%[4,21]。
自2001年PCDH15基因被克隆以来,已有许多研究在USH1F家系中,鉴定出PCDH15的突变位点(表2)。不同家系的临床表型具有相似性,包括先天性双耳对称性感音神经性聋,先天性前庭功能障碍,运动发育迟缓、难以保持平衡。Ahmed等研究发现,USH1F患者独立行走的年纪大多在12-15个月,甚至更晚[7,16]。也有研究报道,患儿36个月时才能独立行走[8]。
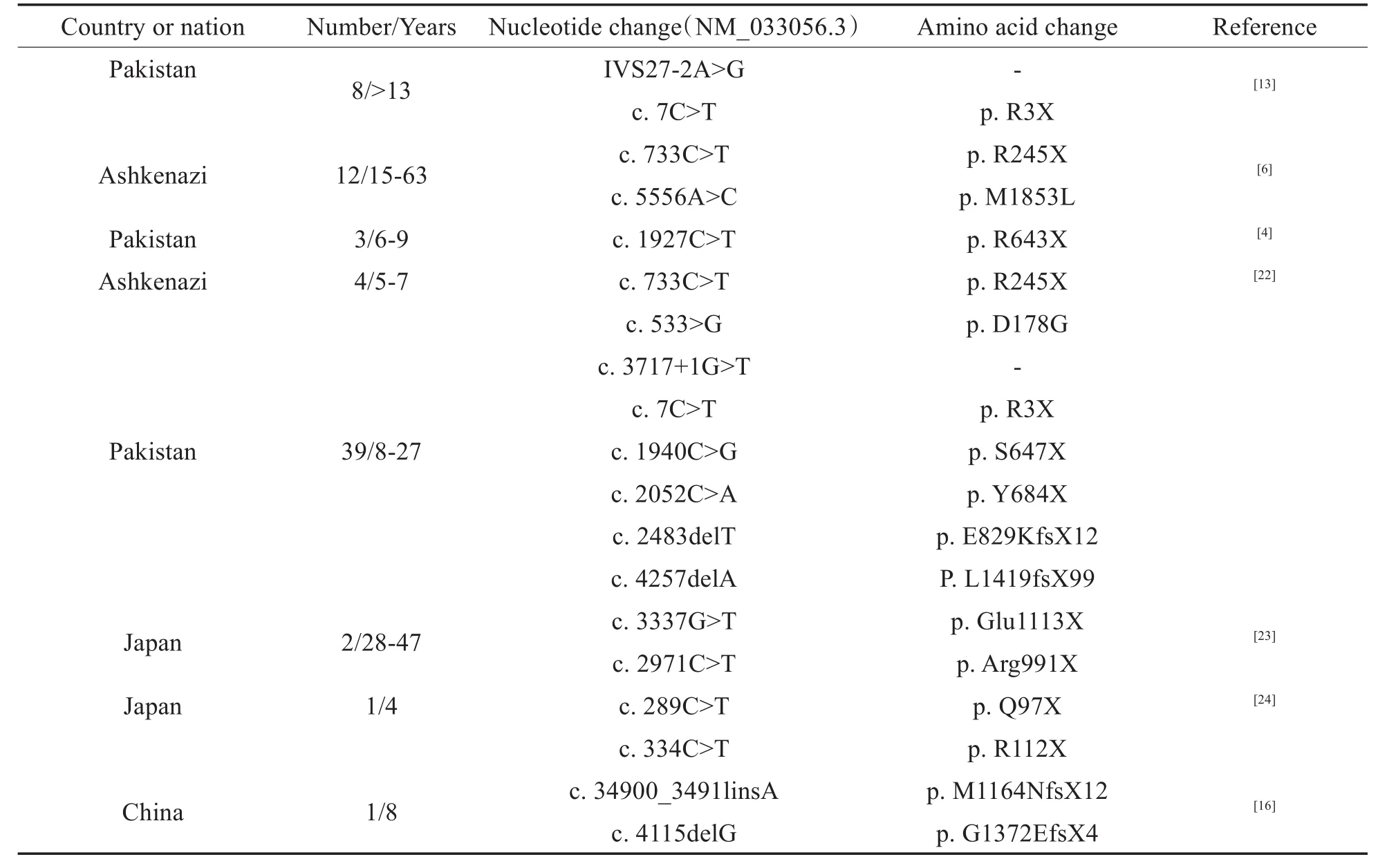
表2 USH1F中的PCDH15突变Table 2 PCDH15 mutation in USH1F families
RP将会在患者青春期前发病,RP典型的检查结果是,眼底血管减弱,视神经顶端呈现蜡状苍白,存在骨针状沉淀物,黄斑部也可能受累[25],确诊RP的主要方法是眼底镜检查与视网膜电图。RP是进行性的,在视觉系统上,视杆细胞最先受到损伤,患者10岁左右将会出现夜盲和隧道视觉等RP早期症状,可能逐渐出现视椎细胞损伤,视野进行性向心性缩小,视觉的对比敏感度、色觉、移动性等功能都将会受到严重影响,最终导致完全失明[26]。在多项研究中,USH1F患者RP确诊年龄大多在10岁之前,也有患者在22岁时才出现RP的报道,目前报道的最小的确诊年龄为2岁[8,25]。除视网膜变性之外,白内障也是常见的特征[27]。所以在USH1F患者眼部表型存在比较大的个体差异。
由于RP多数表现为迟发性,所以仅根据临床表型判断,极有可能将USH1患者诊断为非综合征性耳聋。随着基因检测技术的不断革新,通过新一代测序技术对疾病早期诊断,可为患者提供了许多即时和长期的帮助[28]。2014年,Yoshimura等在日本,首次运用大规模平行测序(massively parallel sequencing,MPS)对17例USH1进行检测,在16例(94.1%)患者中检测到至少携带一个致病性突变[23]。2016年,Yoshimura对227例极重度患儿进行MPS,发现USH1在被诊断为非综合征性耳聋的儿童中的频率为1.3-2.2%[28]。与Sanger测序相比,MPS能够以相对低的成本和较快的速度对整个基因组或目标外显子组的数百万个小片段进行测序[29,30],因此可以快速识别许多致病基因的突变,同时保持成本效益的最大化。此外,对大量基因的同时测序分析有助于发现具有遗传修饰作用或双基因遗传的变异。
Usher综合征在不同种族中常见的致病基因并不相同,在美国和英国白种人群中,MYO7A为最常见的致病基因[31,32],在德系犹太人中PCDH15是最常见的致病基因,PCDH15基因的突变位点c.733C>T(p.R245X,NM_033056.4)在该人群中存在始祖效应,该突变位点占据的比例为50%-60%,p.R245X在德系犹太人中的携带率为0.79%-2.48%[6,22]。也有研究称该突变位点并不局限于德系犹太人,在其他人群中也十分常见[33]。
尽早得到分子诊断对于Usher综合征来说十分重要,通过适当的干预可以延缓视力的减退,例如坚持良好的饮食和生活方式,使用遮阳镜保护眼睛,减少紫外线的伤害[34]。在Vonam等的研究中,运用新一代测序技术,Usher综合征患者的早期诊断年龄通常会早于患者出现视力损失的发病年龄[34]。早期发现进行性视网膜色素变性,在植入人工耳蜗后,儿童可以通过视力阅读唇语,配合听觉康复来获得更好地言语发展。因此早期发现,并在严重失明之前进行眼部手术,对于患儿沟通能力发展至关重要[6,7]。
4 PCHD15基因的特殊突变类型
随着DNA测序技术的不断进步,许多特殊的遗传变异类型也能够被人们发现。技术的不断发展,可以帮助人们丰富对疾病基因型和表型关联的认识。PCDH15基因突变大多是截短突变,该突变破坏基因和蛋白质功能[33]。由于PCDH15存在许多的同工型,不同的同工型在功能上可以互补,第33号外显子主要编码胞体结构域部分,所以有部分发生在第33外显子的截短突变大多没有致病性[33]。
2007年,Le Guedard等首次报道了31例USH1F患者中发现3例患者的PCDH15基因突变存在大片段缺失[8]。PCDH15基因存在大片段重复或者缺失突变的可能性,由于PCDH15基因跨度将近1Mb,开放阅读框为7021bp,前三个外显子覆盖0.42Mb,5’端编码的外显子比例较低,因此极有可能形成许多不同断点的大片段缺失[8]。Vache等在一名法国病人上发现了PCDH15中外显子18-26的杂合缺失,并通过RNA-assay和DNA长阅读测序,在PCDH15内含子序列里发现一段4.6Mb的臂内倒位,最终导致USH1[35]。
CDH23和PCDH15基因编码的蛋白均属于钙粘蛋白超家族,它们是耳蜗静纤毛顶连接的重要组成部分,两者在基因致病突变导致的功能损失和组织学变化具有相似性。2005年,Zheng等发现CDH23和PCDH15基因的突变可以相互作用,出现双基因杂合突变,导致听力损失。他们构建了Cdh23和Pcdh15杂合突变的C57BL/6J小鼠,发现双基因突变小鼠存在更明显的听力损失。另外在三个无关家族中发现CDH23和PCDH15双基因突变导致USH1综合征的遗传证据[36]。除此之外还有许多USH1候选基因中双基因突变的报道,双基因突变患者RP起病年龄更早,表明Usher综合征相关基因之间可能存在修饰作用[23]。
5 小结与展望
PCDH15突变引起DFNB23型和USH1F型遗传性耳聋,均会导致先天性双耳重-极重度耳聋,除此之外,USH1F患者将出现前庭功能障碍以及视网膜色素变性。PCDH15突变在德系犹太人群中存在始祖效应,在中国人群中的发病率尚待进一步统计研究。对于伴有运动发育迟缓或/和夜盲的遗传性耳聋中,要考虑到PCDH15致病的可能性。PCDH15蛋白存在特殊的蛋白质结构域,并且含有众多同工型,导致PCHD15基因可能存在许多截短突变和拷贝数变异。PCDH15存在双基因遗传以及修饰基因的遗传模式,在检测时还需额外关注其他USH1候选基因,这些基因编码的蛋白与PCDH15蛋白有着密切的联系。早期分子诊断有助于疾病的干预和预防,对于患儿未来的言语发展来说至关重要。在未来,运用NGS对遗传性耳聋患者进行突变筛选,不仅可以快速识别致病基因,达到早期诊断、精准治疗的目的,同时还能保持较好的成本效益,有助于遗传咨询、临床指导。并且有助于发现修饰基因或双基因遗传。
除遗传性耳聋之外,噪声性耳聋的病因十分复杂,由遗传因素和环境因素混杂共同作用导致的,有研究表明,在波兰、瑞士、中国人群中,PCDH15基因rs11004085位点的多态性可能是噪声性聋易感的原因之一[37-39],并且PCDH15基因多态位点之间的相互作用可能对噪声性聋易感性有重要影响[40]。因此,提示人们需要考虑到PCDH15基因属于噪声易感基因。
