电压门控钙离子通道药物研究进展
2021-04-13马卓伊戚微岩高新梅徐寒梅胡加亮
马卓伊,戚微岩,高新梅,徐寒梅,胡加亮
(中国药科大学 江苏省合成多肽药物发现与评价中心,江苏 南京 211198)
电压门控离子通道是一类重要的膜蛋白家族,其功能构象对细胞膜电位的变化很敏感,通过允许阳离子通过它们的成孔亚基,再沿电化学梯度向下流动而参与细胞信号传递[1]。电压门控钙离子通道(voltage-gated calcium channels,VGCC)具有共同的架构和3 种与电压相关的不同功能状态:封闭或静息通道、激活或开放通道、失活通道。在去极化时,这些通道以毫秒级的速度由静息状态变为激活状态,然后快速失活,在复极化时返回静息状态[2]。与其他电压门控离子通道不同的是,VGCC 与许多细胞反应有关,包括神经递质释放、肌肉收缩和基因表达。因此,VGCC 能将细胞膜上的电信号转化为细胞内具有重要生理意义的钙离子瞬态[1,3]。
VGCC 是由4 ~ 5 个亚基组成的复合蛋白,这些亚基由多个基因编码,在控制钙离子进出的过程中发挥着不同的作用,根据α1 亚基的差异分为10 个亚型。各种亚型VGCC 在细胞信号传导中发挥着不同的作用:CaV1 亚家族启动神经元的收缩、分泌、调控基因表达和突触的转运和整合;CaV2 亚家族主要负责在快突触中启动突触传递;CaV3 亚家族对于节律放电细胞(如心肌细胞和丘脑神经元)的动作电位的重复放电非常重要[4]。各种亚型VGCC 的功能障碍可能导致从心血管疾病到神经和精神状况的病理、生理学改变,如癫痫、疼痛和自闭症等。
在神经系统中,钙离子通道阻滞剂已被成功用于治疗失神性癫痫,并正在成为疼痛、帕金森病、成瘾和焦虑等疾病的潜在治疗药物。此外,人类钙离子通道基因的突变还与癫痫、偏头痛和共济失调等疾病有关[5]。然而,目前对钙离子通道的认识并不完善,近年来虽然有一些钙离子通道阻滞剂进入临床研究,如CNV-2197944、Z160 等,但进展缓慢,且开发收效甚微[6]。为改善钙离子通道类药物的研发现状,需要对钙离子通道及其作用机制更深入研究,以便开发出更高效、更安全的钙离子通道阻滞剂,更好地造福人类。
1 电压门控钙离子通道
1.1 电压门控钙离子通道亚基的结构和功能
VGCC 中的10 种α1 亚基的4 个主要跨膜区具有相同的跨膜结构,每个跨膜结构有6 个跨膜螺旋(S1、S2、S3、S4、S5 和S6),S1、S2、S3 和S4部分形成了电压敏感域(VSD),带正电荷的S4 部分控制电压依赖的激活,S5 和S6 之间有一个P-loop形成可透过的通路,含有高度保守的带负电荷的氨基酸残基(高压激活型钙离子通道中为谷氨酸)协同形成孔道,对钙、钡、锶等阳离子具有很高的选择性,并且能与非透过的二价阳离子(如镉离子)相互作用。每个跨膜结构包含一个电压传感器基序,在膜去极化时响应使通道打开[7-8]。氨基末端、羧基末端以及4 个跨膜结构之间的链接物面向细胞质,是第二信使调控通道功能的重要靶点[9]。高压激活型钙离子通道的α1 亚基与辅助亚基共同装配后,获得完整的通道功能[4]。
CaVβ 亚 基 有4 个 已 知 编 码 基 因(CaVβ1、CaVβ2、CaVβ3、CaVβ4),同时有多个选择性剪接转录本。CaVβ 是胞质蛋白,与CaVα1 亚基的Ⅰ-Ⅱlinker结构域连接。CaVβ 亚基能够改变CaVα1 的门控特性,对增加细胞表面运输可能起重要作用[10]。CaVα2δ亚 基 有4 种 不 同 的 类 型(CaVα2δ1、CaVα2δ2、CaVα2δ3、CaVα2δ4),作为单独的蛋白转录和翻译,然后裂解成CaVα2和CaVδ,再通过二硫键重新连接[11]。CaVα2 完全暴露在细胞外环境中,CaVδ 的作用相当于一个膜锚,通过一个磷脂酰肌醇锚定在质膜外,CaVα2δ 的共表达能够控制α1 亚基的运输、定位和生物物理特性,并且能够促进加巴喷丁类药物(普瑞巴林和加巴喷丁)的结合,抑制神经病理性疼痛[12]。骨骼肌特异型L-型通道有辅助亚基CaVγ,它有4 个跨膜螺旋,与α1 亚基跨膜螺旋Ⅳ的VSD 区域结合。在神经组织中已经发现了不少于7 个潜在的CaVγ 亚型[13],但关于其功能了解很少(见图1)。

图1 电压门控钙离子通道亚基结构图Figure 1 Subunit structure of voltage-gated calcium channels
1.2 电压门控钙离子通道的分类和亚型
VGCC 按照电压激活特性分类,可分为高电压激活型(high voltage activation,HVA)和低电压激活型(low voltage activation,LVA),HVA 通道在膜去极化程度大时响应激活,而LVA 通道在低电压变化时即可激活,此时的膜电位与神经元静息膜电位相近[14];按照钙离子电流门控特性分类,可分为L-型、P/Q-型、N-型、R-型和T-型;按照α1 亚基的基因型,可分为CaV1、CaV2、CaV3 这3 个家族,每个家族又包含多个成员[15-18](见表1)。
CaVα1 是形成钙离子通道亚型的决定因素。CaV1 通道家族编码3 个不同的L-型通道(CaV1.2、CaV1.3 和CaV1.4)以及一个骨骼肌特异型CaV1.1,表现为激发和持续的电流时间较长,对二氢吡啶(DHP)拮抗剂和激动剂很敏感。CaV2 通道家族包括3 个成员(CaV2.1、CaV2.2 和CaV2.3)。通过选择性剪接和装配特定的辅助亚基,CaV2.1 产生了P-型和Q-型通道,它们均能被ω-agatoxin IVA(从美洲漏斗网蜘蛛毒液中分离的肽)阻断。CaV2.2 编码的N-型通道能够被海洋捕鱼软体动物地纹芋螺Conus geographus和其毒液中分离的ω-芋螺毒素GVIA 和MVIIA 抑制。CaV2.3 对应R-型通道,能被一种从Hyristocrates GigasTarantula 的毒液中分离的多肽SNX-482 抑制[19]。CaV3 通道有3 种类型(CaV3.1、CaV3.2 和CaV3.3),全部属于T-型钙离子通道,通过对被镍离子和钙离子阻断的相对电阻敏感性可以区分CaV3 通道不同类型[20]。
表1 电压门控型钙离子通道的药理和生理特征[15-18]Table 1 Pharmacological and physiological characteristics of voltage-gated calcium channels
2 电压门控钙离子通道上市药物及其作用机制
2.1 L-型电压门控钙离子通道药物
L-型 钙 离 子 通 道(L-type calcium channels,LTCC)是临床上钙离子通道阻滞剂的重要作用靶点,在10 个已知的α1 亚基中,有4 个属于LTCC(CaV1.1、CaV1.2、CaV1.3、CaV1.4),含有二氢吡啶类和其他有机钙离子通道阻滞剂类药物的高亲和力结合位点,但在组织表达和门控特性方面存在差异。CaV1.1 通道几乎只存在于骨骼肌中,CaV1.4 通道主要局限于视网膜中,而CaV1.2 和CaV1.3 在多种组织中有表达,甚至同时存在于同一细胞中[21]。这些通道的异常与一些疾病的发生有关,如精神疾病或心律失常[22]。目前针对LTCC 的药物有二氢吡啶类的伊拉地平(isradipine)、硝苯地平(nifedipine),苯烷胺类的维拉帕米(verapamil)以及苯二氮䓬类的地尔硫䓬(diltiazem)。虽然上述药物的结构不同,但它们均结合在靠近孔道和α1 亚基激活门的一段单独重复的药物结合区域,并与这个结合区域进行立体选择性地可逆结合[23-24]。
2.1.1 二氢吡啶类二氢吡啶类是一类具有广泛生物和药理作用的药物,在心血管疾病的治疗中广泛应用,具有抗高血压、抗心绞痛、扩张血管和抑制心脏等作用,1,4-二氢吡啶环为其基本支架[25]。不带电的二氢吡啶类药物主要稳定和诱导失活的通道状态,它们对失活的LTCC 构象具有很高的亲和力,因此在去极化电压较大时,二氢吡啶类药物阻断心血管LTCC 的半抑制浓度(IC50)很低。二氢吡啶类药物对失活LTCC 的优先亲和性,正解释了其能够在治疗剂量下发挥强大的血管舒张作用的同时,不影响心脏收缩力的特性[26]。
伊拉地平(1)是美国食品和药品管理局(FDA)批准的一种二氢吡啶钙离子通道阻滞剂,可选择性地与大脑海马区CaV1.2 结合,通过抑制钙离子流入细胞质和抑制CaV1.2 的表达来减少β 淀粉样低聚物的毒性,因此可用于治疗阿尔茨海默病。伊拉地平对CaV1.3 也具有很高的亲和力,是一种潜在可行的帕金森病的神经保护剂[27],此外,它对高血压也有很好的治疗作用[28]。硝苯地平(2)能够抑制CaV1.2 通道,阻止钙离子进入细胞内[25],使血管平滑肌得到放松,且作用浓度明显低于对心脏产生显著直接影响时的浓度,可用于治疗心绞痛和高血压[29]。

2.1.2 苯烷胺类和苯二氮䓬类苯烷胺类和苯并噻吩类能够与开放和失活状态的通道高亲和力结合。在生理状态的pH 条件下,苯烷胺类和苯二氮䓬类主要以带正电荷的有机阳离子的形式存在,在通道开放时递送至胞质侧的结合位点[30-31];它们能稳定通道的失活状态,减缓了通道从失活状态中恢复的速度,从而导致频率依赖性的抑制,其主要作用是抗心律失常[32]。
维拉帕米(3)于1962 年由德国Knoll 公司研制成功,商品名为Isoptin,又称为异搏定、异博停、戊脉安,是首个临床上使用的苯烷胺类的抗心律失常、抗心绞痛、抗高血压的药物。维拉帕米属于季苯烷胺类药物,有研究表明,它以不带电的形式穿过细胞膜,进入胞液后,以质子化的形式进入细胞内的孔道(α1)中并与受体位点结合,从而阻断CaV1.2[33]。
地尔硫䓬(4)属于苯二硫氮杂䓬类钙拮抗剂,通常以盐酸盐的形式存在,目前已有100 多个国家在临床上用作抗心绞痛药、降压药。地尔硫䓬能与CaV1.2 的IIIS6 和IVS6 片段结合,阻断钙离子进入细胞,扩张冠状动脉,同时增加冠状动脉血流量,从而使血压和心率降低。与其他药物相比,地尔硫䓬在降压时没有反射性心律加快的不良反应,不增加心肌耗氧量从而保护心脏,在临床使用上更加安全有效[34-35]。
2.2 N-型电压门控钙离子通道药物
N-型(CaV2.2)VGCC 主要表达在神经末梢,控制着神经递质的传递和释放,能够传导电流,调节钙稳态,在传递疼痛信号方面发挥重要作用。N-型VGCC 的抑制剂或调节剂是治疗神经性疼痛的有效药物[36]。
加巴喷丁(gabapentin,5)最初于1993 年在英国获得批准,曾被认为是一种抗癫痫药物,现在也被用作治疗神经性疼痛的一线药物,特别是糖尿病神经痛和疱疹后神经痛。加巴喷丁能够与VGCC的辅助亚基α2δ1 特异性结合,抑制了在背根神经节(dorsal root ganglia,DRG)神经元的突触前末端和背角神经元中,因神经损伤导致的钙离子通道α1 孔道形成单元由细胞质向质膜的运输,也抑制了α2δ1 从DRG 到背角神经元的轴向顺行运输[37-38]。普瑞巴林(pregabalin,6)是第2 代靶向α2δ1 的药物,于2004年在欧洲被批准用于外周神经性疼痛的控制,并作为局部癫痫的辅助治疗[39]。普瑞巴林可能是通过下调脊髓瞬时阳离子通道亚家V 成员1 蛋白受体(the transient receptor potential cation channel subfamily V member 1,TRPV1)的表达来缓解神经病理性疼痛[40],是治疗带状疱疹后神经痛的一线药物。

齐考诺肽(ziconotide,商品名Prialt,见图2)于2004 年在FDA 和欧盟获得批准,是人工合成的ω-芋螺毒素MVIIA,能与CaV2.2 通道的特异性可逆结合,通过阻断脊髓背角传入神经释放前痛觉神经递质而产生镇痛作用[41],目前用于治疗阿片药物治疗无效的慢性疼痛。齐考诺肽是由25 个氨基酸组成的多肽,给药方式为鞘内注射。与阿片类药物吗啡相比,使用齐考诺肽不会产生急性戒断症状,也没有任何已知的上瘾风险,长时间服用也不易产生耐受[42-43]。如果需要停止使用齐考诺肽也可以立即停止,无需减少剂量[44]。齐考诺肽的这些优点,使其成为治疗慢性疼痛,尤其是对吗啡耐受疼痛的重要药物,也为开发新的镇痛药物提供了思路。
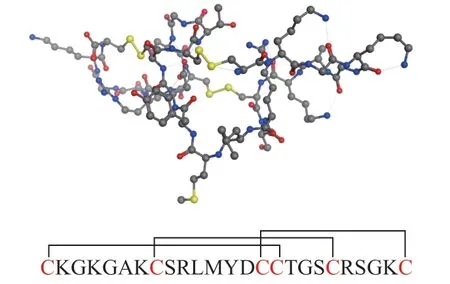
图2 齐考诺肽的球棍模型结构图Figure 2 Ball-and-stick model of ziconotide
2.3 R-型电压门控钙离子通道药物
R-型VGCC 属于钙离子通道CaV2.3 亚型,它的作用至今尚未明确。SNX-482 是一种选择性的、电压依赖的CaV2.3 通道抑制剂,它能与CaV2.3 的结构域Ⅲ和Ⅳ相互作用,有效阻滞R-型VGCC,加大剂量后也可以阻滞L-型和P/Q-型VGCC 和钠离子通道[45]。
3.4 T-型电压门控钙离子通道药物
T-型VGCC 为钙离子通道CaV3.2 亚型,是传入性疼痛通路中疼痛信号的重要调控因子,在不同的慢性疼痛状态下,钙离子通道的活性受到不同程度的调控。因此,抑制DRG 神经元和脊髓背角的T-型VGCC 可以作为镇痛的靶点。T-型VGCC 在膜去极化程度较小时打开,允许钙离子进入细胞,调节神经元的兴奋性,促进脊髓背角中神经递质的释放。参与疼痛信号传导的主要CaV3 通道亚型是CaV3.2[46]。
乙琥胺(ethosuximide,7)是T- 型VGCC 阻滞剂,临床上用于治疗失神性癫痫。乙琥胺也可以逆转动物模型中的疼痛,但还没有在人体中得到有效性的报道[47]。咪拉地尔(mibefradil,8)也是T-型VGCC 阻滞剂,且对CaV3.2 的阻断强于CaV3.1和CaV3.3,可使神经损伤引起的疼痛和神经兴奋恢复正常,在临床上已被用于治疗慢性心绞痛和高血压[48],但是由于严重的药物相互作用,咪拉地尔上市一年后被撤市,10 年后被FDA 批准为孤儿药,用于研究其对脑癌、卵巢癌和胰腺癌的疗效[49]。目前最有前景的T-型VGCC 阻滞剂是Z944(9)和ACT-709478(10),已经进入临床试验阶段[50]。
2.5 P/Q-型电压门控钙离子通道药物
P/Q- 型VGCC 由基因CACNA1A编码,选择性剪接来自α1 亚基基因的信使mRNA 产生了P-型和Q-型电流(分别称为CaV2.1a 和CaV2.1b),这些变异体可以通过对蜘蛛毒素ω-agatoxin IVA 的敏感性进行区分[51]。与P-型VGCC 相比,Q-型VGCC 失活慢,这是因为CaV2.1b 的Ⅰ-Ⅱlinker 结构域上插入了一个氨基酸(缬氨酸)。CaV2.1 是大脑中最丰富的α1 亚基,在大鼠和人类的肾脏中也有表达[52]。
蜘蛛毒素ω-agatoxin IVA 是一种选择性抑制P/Q-型VGCC 的多肽,它能够在低于10 nmol · L-1的浓度下阻滞P-型VGCC,而在较高的浓度下阻滞Q-型VGCC,但是由于它的相对分子质量较大,不适合作为临床药物使用[53]。

3 结语
VGCC 不仅能够改变细胞电位,还能进行信号转导,这赋予了VGCC 更加广泛的功能。近年来,随着实验技术的提高和研究手段的改进,VGCC 的结构得到了进一步解析,这促进了VGCC 阻滞剂的发现及进入临床应用。L-型VGCC 在抗高血压、抗心律失常和抗心绞痛方面发挥重要作用,L-型VGCC阻滞剂如二氢吡啶类的硝苯地平、伊拉地平,苯烷胺类的维拉帕米,苯二硫氮杂䓬类的地尔硫䓬等,均在临床上得到了广泛的应用[27,29,33-35]。N-型和T-型VGCC 阻滞剂与镇痛关系密切,如加巴喷丁和普瑞巴林[12];近年来对动物毒素的研究越来越多,动物毒性肽如齐考诺肽的上市为开发此类药物提供了新的思路[41-44];此外,甚至发现了T-型VGCC 的抗癌作用[54]。R-型和P/Q-型VGCC 的研究较少,对其功能的了解尚不完善,目前也没有相关药物上市。本文对上述VGCC 的结构、作用靶点和上市药物进行了概述(见表2),期望为其后续研发提供参考。
笔者所在课题组从齐考诺肽的发现和应用中受到启发,选择了少棘蜈蚣(Scoropendra subspinipes mutilans)这一传统中药材,通过构建cDNA 文库,使用高通量测序技术测序后,在得到的蛋白质序列中筛选具有镇痛活性的多肽并进行活性研究,并且已经发现了具有一定活性的多肽片段。目前正在对这些多肽片段进行进一步的活性评价,期待它们能表现出良好的镇痛效果。

表2 电压门控型钙离子通道药物的靶点和适应证Table 2 Targets and indications of voltage-gated calcium ion channel drugs
