基因多态性与药物不良反应发生风险的相关性及其临床证据
2021-04-13李玉娇初亚男黄晓晖张婕妤周国华
李玉娇,初亚男,黄晓晖,张婕妤,周国华
(东部战区总医院临床药学科,江苏 南京 210002)
药物不良反应(ADR)是指合格药品在正常用法用量下出现的与用药目的无关的有害反应,是临床实践和制药业关注的主要问题之一[1]。2019 年全国ADR 检测网络收到“药品不良反应/事件报告表”151.4 万份,较2018 年增长了1%;其中严重药品不良反应/事件报告15.6 万份,占同期报告总数的10.3%[2]。在美国,约6.7%的住院患者出现严重的ADR,ADR 是仅次于癌症、心脏病和休克的第四大死亡原因[3]。导致ADR 发生的因素有很多,包括合并用药、生活方式、年龄和饮食等。越来越多的研究表明,ADR 和基因多态性关系密切。药物基因组学对药物疗效和安全性的影响概率高达80%,超过600 个基因与药物疗效有关,约200 个药物基因与ADR 相关[1]。体内参与药物吸收和处置的药物代谢酶、转运蛋白及相关受体基因多态性的改变是导致药物反应个体差异的决定性因素[4-5]。随着人类基因组学和药物基因组学研究的不断深入,识别药物个体差异的相关基因及其遗传变异和ADR 间的临床联系不断被证实,部分研究结果已形成临床指南[6],这为实现患者个体化药物治疗、避免或减少遗传变异引起的ADR 提供了可能。
1 基因多态性可预测ADR 风险
目前,药物基因组学在临床ADR 预测中的应用需求越来越大。对患者先行药物相关基因检测,可以有效降低基因多态性相关ADR 的发生风险。遗传药理学和药物基因组学知识库(PharmGKB)是一个收集、指导与传播关于临床可操作基因-药物ADR 关联和基因型-表型关系等信息证据的药物基因组学数据库。PharmGKB 使用一个简单的6 级证据强度评估系统(见图1、表1)来评价遗传变异与ADR 之间关联证据的强度。目前为止,至少有469种药物相关基因被证实与226 种临床药物的ADR有关,证据强度从4 级(弱)到1A 级(强)不等,主要涉及抗肿瘤药物、中枢神经系统药物及心血管系统药物等。
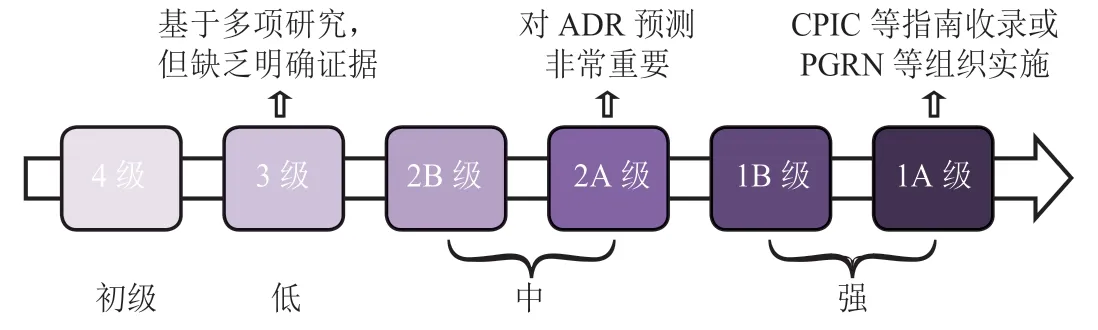
图1 PharmGKB 用于评价药物基因临床证据强度的6 级评分系统Figure 1 PharmGKB 6-level scoring system for the strength of clinical evidence of a pharmacogene

表1 PharmGKB 数据库定义的遗传变异与ADR 风险间的关联强度Table 1 Strength of evidence for the association between genetic variations and the risk of ADR defined by PharmGKB
截 至2020 年5 月,PharmGKB 数 据 库 收 录ADR 相关的临床证据强度为1A 级的药物基因有13 个,包括细胞色素P450 2C9(CYP2C9)、细胞色素P450 2C19(CYP2C19)、细胞色素P450 2D6(CYP2D6)、二氢嘧啶脱氢酶(DPYD)、葡萄糖-6-磷酸脱氢酶(G6PD)、巯基嘌呤甲基转移酶(TPMT)、尿苷二磷酸葡糖醛酸转移酶1A(UGT1A1)、L型钙离子通道α1S亚单位(CACNA1S)、三磷酸核苷酸二磷酸酶(NUDT15)、人类白细胞抗原A(HLA-A)、人类白细胞抗原B(HLA-B)、人兰尼定受体1(RYR1)和有机阴离子转运多肽1B1(SLCO1B1)。与ADR 相关的临床证据强度为1B级的药物基因有4 个,分别为CYP2D6、G6PD、线粒体编码12S RNA(MT-RNR1)和着色性干皮病基因组C(XPC),它们的遗传变异与ADR 风险关系较大。这些重要的药物相关基因已被CPIC 和荷兰皇家药剂促进协会-药物遗传学工作组(DPWG)等权威机构发布的指南收录,或由PGRN 等卫生系统实施临床实践。此外,有22 种药物基因与临床证据强度2A 或2B 的中度ADR 风险有关,约有443 种药物基因与ADR 的相关性只具有较低水平(3 ~ 4 级)的临床证据。截至2020 年12 月,CPIC 共制定了25 个药物指南,包括66 对基因-药物信息,并将药物-基因关联的使用推荐强度分为4 个等级(A、B、C 和D),其证据强度评估标准与PharmGKB 有很高的一致性。
另外,美国食品和药品管理局(FDA)列出了至少50 个基因作为临床药物基因组生物标志物,这些基因与超过145 种药物的药物反应、药物清除和ADR 风险显著相关,在识别患者对药物的反应、优化药物剂量以及避免遗传变异引起的ADR中发挥了重要作用。截至2020 年2 月,至少有16个与ADR 风险有关的基因已经被FDA 列为药物基因组生物标志物,包括HLA-A、HLA-B、N-乙酰化转移酶1(NAT1)、N-乙酰化转移酶2(NAT2)、CYP2D6、TPMT、NUDT15、UGT1A1、CYP2C19、DPYD、CYP2C9、血清胆碱酯(BCHE)、细胞色素P450 2B6(CYP2B6)、人类白细胞抗原DRB1(HLA-DRB1)、人类白细胞抗原DQA1(HLA-DQA1)和SLCO1B1[7],其中14 个基因也被PharmGKB 数据库收录。FDA 强调药物遗传信息在药品标签中的应用,并提供有充分临床证据支持的药物遗传学关联,FDA 和PharmGKB 为帮助临床医生根据患者基因型确定治疗策略、药物合适剂量或疗效和毒性的评估提供了可能性。目前,主要的具有高级证据等级的ADR 风险相关的基因-药物信息见表2。

表2 具有高级证据等级的 ADR 风险相关的基因-药物信息Table 2 Gene-drug information with advanced clinical evidence related to ADR risk
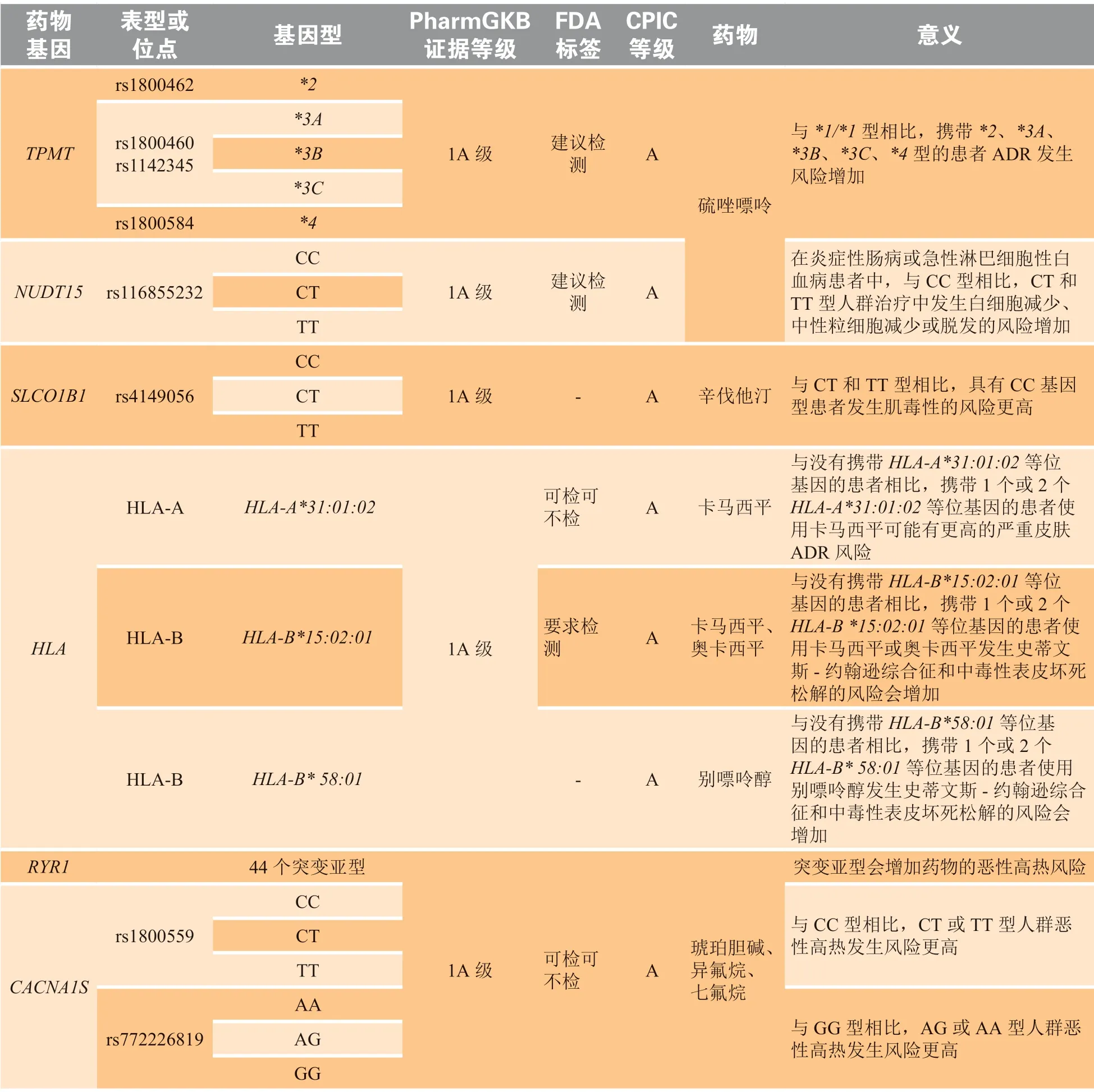
续表2
2 基因多态性与ADR 的临床相关性
患者年龄、性别、器官功能、精神状态和基础疾病状况等众多非基因因素均会影响个体的药物反应[8]。越来越多的临床证据显示,药物代谢酶、转运蛋白、靶受体和其他作用靶点的遗传变异会影响药物在患者体内的药物代谢动力学/药效学(PK/PD)过程[9],是造成药物效应个体差异和ADR 风险的重要原因。
2.1 代谢酶基因多态性与ADR 的临床相关性
药物及其活性代谢物在体内通过生物转化为代谢物排出体外,体内的药物代谢酶分为可参与Ⅰ相生物转化的CYP450 酶系,包括CYP2C9、CYP2C19、CYP2D6 等,参与Ⅱ相生物转化的DPYD、UGT1A1、TPMT、G6PD 等[10]。代谢酶的活性由一对等位基因决定,慢代谢型(PM)携带2个无功能的等位基因;中间代谢型(IM)携带2 个活性降低的等位基因;快代谢型(EM)携带一对或一个功能正常的等位基因;超快代谢型(UM)携带2个以上的活性基因[11]。药物代谢酶能代谢95%以上的临床使用药物,其多态性决定了药物治疗效果的个体差异和ADR 的发生风险。
FDA 药物标签和PharmGKB 数据库中有充分的1A 和1B 级临床证据明确了Ⅰ相代谢酶CYP450 变体与临床药物ADR 的风险高度相关:1)CYP2C9是CYP450 家族的重要成员之一,主要分布在肝脏组织,代谢约10%的临床常用药物,其中包括芳香类抗癫痫药(如苯妥英)和抗凝药(如华法林)。CYP2C9*2和CYP2C9*3是最常见的2 种缺陷等位基因,在中国人群中的分布频率分别为0%和10%[11]。CYP2C9和维生素K 环氧化物还原酶复合体1(VKORC1)的遗传多态性与华法林和苯妥英的ADR 有关[12]。CYP2C9*2可使S 型华法林代谢清除率降低30% ~ 40%,而CYP2C9*3则可使其降低80% ~ 90%,导致华法林在体内蓄积,从而增加出血风险[13-14]。VKORC1 是华法林靶标酶,1639G > A 突变会降低VKORC1 酶的蛋白表达水平,影响华法林治疗的敏感性[15]。FDA(2017年)批准的华法林药物标签提示,与CYP2C9野生 型 患 者 相 比,CYP2C9*1/*3、CYP2C9*2/*2、CYP2C9*2/*3、CYP2C9*3/*3基因型患者需要更长时间才能达到给定给药方案的最大国际标准化比值(INR),CYP2C9和VKORC1基因型决定了华法林的药物反应和ADR 风险。CPIC 指南(2017 年)和DPWG 指南(2017 年)也建议在使用经过验证的药物遗传算法计算华法林用量的基础上,减少CYP2C9*2、CYP2C9*3和 携 带2 个VKORC1等 位基因变体(1639G > A)基因型患者的华法林使用剂量。一项对布-加综合征(BCS)患者华法林抗凝治疗出血事件的研究显示,CYP2C9*1、CYP2C9*2、CYP2C9*3和VKORC1(1639G > A)的单倍体突变会增加BCS 患者华法林治疗的出血发生风险[16]。另有一项在东亚癫痫患者中对基因多态性与苯妥英毒性及疗效的研究显示,CYP2C9*3型可能会增加苯妥英引起的严重皮肤不良反应(SCAR)的发生风险[17]。2)CYP2C19 是抗血小板药物(如氯吡格雷)、质子泵抑制剂(如奥美拉唑)、抗抑郁药(如西酞普兰)和抗真菌药(如伏立康唑)的主要代谢酶。CYP2C19*2和CYP2C19*3是中国人群的2 种主要缺陷等位基因,发生频率分别为23% ~ 35%和2% ~7%,突变型为PM;CYP2C19*17在中国人群的发生频率为0.5%~4%,编码的CYP2C19 酶转录活性增强,突变型为UM[18]。氯吡格雷为一种无药理活性的前体药物,经CYP2C19 作用后的活性代谢产物与血小板表面的P2Y12 受体结合,抑制二磷酸腺苷(ADP)介导的血小板活化和聚集,发挥抗血小板效应[19]。研究证实,氯吡格雷的ADR 与携带CYP2C19*2/*2、CYP2C19*2/*3、CYP2C19*2/*4、CYP2C19*2/*5、CYP2C19*2/*8和CYP2C19*3/*3的PM 之间存在很强的相关性[20]。FDA(2017 年)批准的氯吡格雷药物标签显示,携带2 个CYP2C19突变等位基因的患者抗血小板凝聚作用减弱,建议使用氯吡格雷前检测CYP2C19基因型,对于氯吡格雷PM 患者,推荐使用另一种血小板P2Y12 抑制剂。CPIC 指南(2013 年)和DPWG 指南(2017年)也建议对于正在接受经皮冠状动脉介入(PCI)治疗的急性冠脉综合征(ACS)患者,在没有禁忌症的情况下,针对CYP2C19的EM 或IM 患者可选择其他抗血小板治疗(如普拉格雷和替格瑞洛)[19]或将氯吡格雷的剂量增至150 mg · d-1或600 mg(负荷剂量)以避免支架栓塞和心肌梗死等心血管风险。一项对青少年焦虑抑郁症接受西酞普兰治疗的研究发现,与CYP2C19的 EM 人群相比,CYP2C19的PM 人群对西酞普兰的耐受性更差,ADR 发生更多[21]。此外,曲洪澜等[22]对侵袭性真菌感染患者CYP2C19基因多态性与PK 关系的研究显示,CYP2C19*1/*2、CYP2C19*1/*3、CYP2C19*2/*2、CYP2C19*2/*3和CYP2C19*3/*3基因型人群出现血清伏立康唑水平过高、神经毒性等ADR 的概率更大。FDA(2019 年)对伏立康唑的药物标签显示,CYP2C19的PM 和IM 患者的伏立康唑暴露量分别是正常代谢者的4 倍和2 倍。CPIC 指南(2016 年)和DPWG 指南(2019 年)建议CYP2C19的PM 患者慎用伏立康唑,建议选用伊莎芙康唑、脂质体两性霉素B 和泊沙康唑等其他抗真菌治疗方案或使用时仔细监测伏立康唑血药浓度[23]。3)CYP2D6 是一种高度基因多态性的药物代谢酶,有超过104 个不同的变体,与多种药物反应有关。CYP2D6*3、CYP2D6*4、CYP2D6*5和CYP2D6*6是最常见的突变等位基因,与抗抑郁药物[24-26](如阿米替林、去甲三嗪和帕罗西汀等)、中枢镇痛药物[27-29](如可待因、曲马多和氧可酮等)和抗肿瘤药物[30](如三苯氧胺)的ADR 有关,发生心脏毒性、神经毒性、呼吸系统毒性和胃肠道毒性的风险均较高。研究显示,82%的携带一个功能性CYP2D6等位基因的抑郁症患者(n= 50),阿米替林治疗后出现明显的心脏和神经毒性[31]。一项基于92 例乳腺癌患者的研究发现,CYP2D6 代谢异常的癌症患者更容易出现他莫西芬诱导的子宫内增生等ADR[32]。
PharmGKB 的1A 和1B 级临床数据显示,包括DPYD、UGT1A1、TPMT 和G6PD 的Ⅱ相代谢酶是影响药物ADR 风险的重要因素。1)DPYD编码的脱氧吡啶啉(DPD)是结肠癌、直肠癌、乳腺癌、胃癌、胰腺癌化疗药——氟嘧啶类药物(5-氟尿嘧啶和卡培他滨等)分解代谢的限速酶。10% ~30%的患者在使用氟嘧啶治疗早期出现腹泻、黏膜炎及中性粒细胞减少症等严重的、可能危及生命的毒性反应[33]。DPYD根据突变剪切位点不同可分为近40 个不同变体,其中DPYD*2A、DPYD*13、DPYDrs67376798 和DPYDrs75017182 是 与 氟 嘧啶ADR 最相关的突变等位基因[34]。DPD 失活可导致酶活性降低,氟嘧啶代谢障碍,使其分解减少而合成增多,细胞毒性显著增强,引发胃肠道反应、骨髓抑制和神经系统毒性等[35-36]。一项纳入2 038例癌症患者的DPYD*2A基因多态性前瞻性研究显示,降低DPYD*2A突变患者的氟嘧啶类药物治疗剂量可将ADR 的发生率从73%降至28%,进行DPYD*2A基因多态性筛查的患者的平均治疗费用显著减少[37]。FDA 于2019 年更新的氟尿嘧啶药物标签表明,5-氟尿嘧啶相关的罕见的、严重ADR(如口炎、腹泻、中性粒细胞减少和神经毒性)与缺乏DPD 活性有关。CPIC 指南(2013 年)建议根据DPYD基因型调整氟嘧啶(卡培他滨、氟尿嘧啶和替加氟)的用药剂量,对携带2 种无功能DPYD变 异(DPYD*2A和DPYD*13)的PM 患者使用替代药物或将氟嘧啶的起始剂量降低50%。2)UGT1A1*28和UGT1A1*6是2 种最常见的缺陷型基因,在汉族人群中的发生频率分别为15% ~ 30%和13% ~ 33%[38]。UGT1A1*28纯合突变型HIV 患者服用阿扎那韦后患高胆固醇血症的ADR 可能性较UGT1A1*1型明显增加[39]。此外,UGT1A1基因多态性与伊立替康诱导的ADR 之间有很强的相关性。伊立替康在体内由羧酸酯酶(CES)水解转化成具有抗肿瘤活性的7-乙基-10-羟基喜树碱(SN-38);SN-38 在肝脏中由UGT1A1 代谢为无活性的SN38G后经尿液及胆汁排出体外,因此,UGT1A1 是代谢伊立替康活性产物的关键酶,其基因多态性是引起剂量限制性毒性反应的重要原因[40]。周琰等[38]对82 例结直肠癌患者UGT1A1基因多态性与伊立替康化疗ADR 间关系的研究发现,UGT1A1*28携带者中性粒细胞数量减少,骨髓抑制性ADR 的发生概率显著增加,而UGT1A1*6基因型和伊立替康相关ADR 无明显相关性。Yang 等[41]研究也表明,在亚洲人群中,UGT1A1*28突变型癌症患者发生伊立替康诱导的严重粒细胞减少和腹泻的风险增加。3)TPMT 是一种催化巯嘌呤类药物(如6-巯基嘌呤)的胞质酶。巯嘌呤在肝脏内经TPMT 代谢为无活性的6-甲基巯嘌呤(6-MP),并干扰活性产物6-硫鸟苷酸(6-TNG)的合成。TPMT遗传多态性与巯嘌呤类药物相关的严重造血毒性之间有很强的相关性[42]。TPMT*2、TPMT *3A、TPMT *3B、TPMT *3C是最常见的TPMT缺陷变体[43],其携带者TPMT 活性降低,体内6-TNG 浓度升高,骨髓抑制风险增加[44]。4)G6PD 缺乏症是一种最常见的由X 染色体上的显性基因控制的遗传性酶病,男性患者的急性溶血风险更高[45]。该症多发于非洲、东南亚、地中海沿岸及我国,中国人群的发生频率为4.5%[46]。研究发现,拉布立酶可用于预防血液恶性肿瘤患者的急性高尿酸血症,但G6PD 缺乏会增大用药时高铁血红蛋白血症的发生风险,因此,在给G6PD 缺乏症患者使用拉布立酶时需考虑风险和收益[47]。另一项对212 例刚果发热儿童的G6PD基因多态性与ADR 及预后关系的研究发现,抗疟疾药物(如伯氨喹和氯丙胍氯苯砜青蒿琥醑)的急性溶血性贫血风险与G6PDrs1050828 基因缺陷相关[48]。因此,在使用抗疟疾药物前检测G6PD基因型可规避疟疾防控过程中的溶血风险,减少相关ADR。5)亚甲基四氢叶酸还原酶(MTHFR)是叶酸代谢的限速酶,在核苷酸合成中起重要作用。MTHFRrs1801133、MTHFRrs1801131 位点突变与患者服用环磷酰胺、氟尿嘧啶和甲氨蝶呤的ADR 相关[49-50]。MTHFRC677T和MTHFRA1298C 变体是导致酶活性降低的最常见2 种功能多态性基因,一项Meta 分析显示,MTHFRC677T 可导致类风湿性关节炎患者甲氨蝶呤中毒,表现为转氨酶增加及口腔炎、恶心、脱发和皮疹等ADR[51]。同时,MTHFRC677T 多态性可升高血浆同型半胱氨酸水平,与胃肠道症状等ADR 有关[52]。
2.2 转运蛋白基因多态性与ADR 的临床相关性
位于细胞质膜上的药物转运蛋白在药物的吸收、分布和排泄过程中发挥重要作用,主要分为介导药物进入细胞内的摄取转运体和分泌药物到细胞外的外排泵两大类。转运蛋白的基因多态性是决定药物反应和ADR 风险的关键因素之一[53-54]。迄今为止,许多药物转运蛋白的外显子变体,如P-糖蛋白(P-pg)(2B 级)和内流转运溶质载体(SLCs)(1A级)已被广泛应用于临床实践。ABCB1编码的P-pg是一种广泛分布于肾小管和肿瘤组织的外排性转运体,能够转运包括心血管药物、抗肿瘤药物和抗菌药物等结构不同的化合物。肾移植供体肾小管上皮细胞中P-pg 低表达与环孢素引起的肾毒性密切相关。Sallustio 等[55]研究表明,与ABCB1G1199A 亚型相比,ABCB1C3435T 亚型使肾移植患者环孢素的肾脏清除率降低约25%,由于血药浓度上升,使肾脏毒性风险增加。另一项基于10 932 个受试志愿者的研究发现,ABCB1C3435T 亚型显著影响地高辛的药物反应,导致地高辛转运障碍,血清浓度升高,ADR 发生风险增大[56]。此外,临床证据强度为3 ~ 4级的ABCB1其他变体,如rs1045642、rs2032582、rs2032582、rs1128503、rs2235040 和rs2238476 也 与药物ADR 有关[57],但仍需通过充分的临床研究来证实。SLCO1B1编码的有机阴离子转运多肽(OATP1B1)是一种摄取转运体,SLCO1B1突变导致OATP1B1 合成受阻,可使血中他汀类药物浓度升高,肌病发生的风险增加。研究已经证实,SLCO1B1rs4149056 C 等位基因与阿托伐他汀、氟伐他汀、洛伐他汀、普拉伐他汀、瑞舒伐他汀和辛伐他汀等他汀类药物引起的肌病相关,而SLCO1B1rs2306283(c.388A > G)G 等位基因则可以抵消SLCO1B1rs4149056 突变引起的他汀类相关肌病ADR[58]。
2.3 药物靶点基因多态性与ADR 的临床相关性
药物靶点特别是代谢通路中限速酶的基因多态性对药物作用和ADR 有非常大的影响。约50%的药物通过与膜受体结合发挥治疗作用,因此药物受体的遗传变异是影响药物反应的关键因素。到目前为止,大量与ADR 相关药物受体的基因多态性被PharmGKB 收录,如μ-阿片受体和β2-肾上腺素能受体。1)OPRM1编码的μ-阿片受体是最常见的阿片受体之一,也是内源性和外源性阿片类镇痛药物(如吗啡和芬太尼)的主要作用靶点[59]。人μ-阿片受体的基因多态性研究主要集中在rs1799971(118A > G)上,118A > G 突变可导致μ-阿片受体的活性降低,影响阿片类镇痛药物的敏感性和治疗效果,容易出现阿片类药物成瘾等ADR[60-61]。2)β2-肾上腺素能受体(ADRB2)的基因多态性是决定β2 受体激动剂(如沙丁胺醇、福莫特罗等)气道反应性的关键因素[62]。ADRB2R16G 和ADRB2Q27E 位点突变显著影响ADRB2 受体功能,导致β2 受体激动剂与ADRB2 结合异常,降低药效并增加ADR 风险[63]。β2-肾上腺素能受体的基因多态性与哮喘表型、支气管亢进以及急慢性β 受体激动剂的治疗效果有关,T164I 位点突变会影响平滑肌β2 受体腺苷酸环化酶的偶联,从而减弱支气管药沙丁胺醇的反应性[64]。因此,鉴定药物受体的基因型有利于了解受体的结构与功能,实现个体化给药治疗,降低甚至避免ADR。
2.4 免疫分子基因多态性与ADR 的临床相关性
HLA 位点是与ADR 相关的最重要的药物基因组学生物标志物之一[65]。HLA 复合物是免疫相关基因家族的重要成员,位于人第六号染色体短臂,由100 余个紧密连锁的基因座组成,具有高度基因多态性。按HLA 产物的分布、结构和功能可以分为3 类,Ⅰ类抗原包括HLA-A、HLA-B 及HLA-C,Ⅱ类抗原包括HLA-DP、HLA-DQ 及HLA-DR,Ⅲ类抗原位于上述2 类抗原区域之间,包括编码热休克蛋白(HSP)、肿瘤坏死因子(TNF)和补体C2、补体C4 等可溶性蛋白的基因。PharmGKB 1A 级临床证据显示,HLA-B*57:01与氟氯西林诱导的肝毒性和核苷类抗逆转录药物(阿巴卡韦和奈韦拉平)诱导的超敏反应有关,HLA-DRB1*15:01和HLA-DQB1*06:02位点突变与非甾体抗炎药罗美昔布引起的药源性肝损伤有关,HLA-A*31:01和HLA-B*15:02与抗癫痫药物卡马西平所致超敏反应和SCAR 存在关联[66]。几乎仅特定的亚裔人群携带HLA-B*15:02和HLA-A*31:01的等位基因,突变频率约为10% ~ 15%[67]。FDA(2018 年)批准的卡马西平药物标签包含对HLA-B*15:02和HLA-A*31:01基因型癫痫患者卡马西平治疗的黑框警告,在卡马西平治疗前,应对所有HLA突变的风险人群癫痫患者进行HLA-B*15:02和HLA-A*31:01基因检测。CPIC 指南(2016 年)和DPWG 指南(2017 年)也建议未使用过卡马西平的HLA-B*15:02阳性、HLA-A*31:01任何基因型或未知基因型的癫痫患者不使用卡马西平,使用替代药物治疗,以规避史蒂文斯-约翰逊综合征(SJS)/中毒性表皮坏死松解症(TEN)等严重皮肤超敏反应的发生风险。基于日本和泰国等亚洲人群患者抗癫痫治疗的队列研究显示,对HLA-A*31:01和HLA-B*15:02基因筛查阳性的癫痫患者,选用卡马西平替代药物可显著降低卡马西平诱导的皮肤ADR 发病率[68-69],表明HLA基因检测在癫痫治疗的临床实践中具有可行性。综上,HLA等位基因与药源性ADR 风险的相关性是HLA研究领域的一个新应用,具有重要的临床意义。
2.5 其他分子基因多态性与ADR 的临床相关性
体内参与药物吸收和处置的其他分子的基因多态性也会导致药物反应个体差异和ADR 的发生。1)CACNA1C编码的Cav1.2 L-型电压门控钙通道(LTCC)α1C 亚单位和RYR1编码的钙离子通道蛋白兰尼定受体1(RyR1)主要分布于心肌细胞,是介导钙离子内流的重要途经,发挥维持正常心脏功能的重要作用。CACNA1C基因的rs1800559 位点T等位基因、rs772226819 位点AG 型及RYR1基因中的44 个突变亚型均会增加琥珀胆碱、异氟烷等肌松麻醉药物的恶性高热风险[70]。2)NUDT15 为一种核苷二磷酸酶,可催化巯嘌呤药物的细胞毒性代谢物三磷酸硫鸟嘌呤(TGTP)转化为毒性较低的单磷酸硫鸟嘌呤。NUDT15突变可导致过量TGTP 掺入DNA 双链,造成DNA 损伤和细胞凋亡,加重巯嘌呤药物的细胞毒性[71]。R139C等位基因是首个被发现与巯嘌呤骨髓抑制毒性有关的NUDT15突变位点,在欧洲和非洲人群中突变频率小于1%,在亚洲人群的突变频率最高,约为9.5%[72]。这种单核苷酸多态性(SNP)引起的氨基酸变化可导致蛋白质稳定性和酶活性降低,携带该等位基因的患者表现出严重的骨髓抑制反应。基于慢性肠道炎症性疾病(IBD)[73]和欧洲急性淋巴性白血病(ALL)患者[74]的研究证实,NUDT15R139C 与服用巯嘌呤药物(AZA 和6-MP)导致的以白细胞和中性粒细胞减少为特征的骨髓毒性有关。2018 年CPIC 也更新了基于TPMT和NUDT15基因型的巯嘌呤药物使用剂量指南,建议NUDT15*1/*2、NUDT15*1/*3、NUDT15*2/*5、NUDT15*3/*6基因型的IM 患者和NUDT15*2/*2、NUDT15*2/*3、NUDT15*3/*3基 因 型 的PM 患 者降低硫唑嘌呤、巯基嘌呤和硫鸟嘌呤的起始剂量或使用非硫代嘌呤免疫抑制剂治疗,以期减少相关ADR 的发生风险[44]。NUDT15的其他罕见突变,如NUDT15*4、NUDT15*5、NUDT15*6、NUDT15*7、NUDT15*8、NUDT15*9亚型与巯嘌呤毒性的相关性目前仅局限于体外实验,仍需充分的临床实践来确认。
3 基因检测在预测和减少ADR 中的应用
ADR 是临床药物治疗中经常出现的难题,往往只能在发生后处理而不能提前预测,但是药物基因组学的发展为解决这一困境提供了可能性。在用药前预先对相关基因进行检测,依据基因表型通过替换药物、升高或降低给药剂量,可以有效减少或避免由于个体化基因差异造成的ADR 事件的发生。同时,当ADR 发生时,除了从特殊人群、超剂量使用、不当配伍用药、给药时机等非基因层面对ADR原因进行分析外,还可以根据PharmGKB 等数据库中提供的基因信息及时调整剂量或更换药物。因此,药物ADR 相关的基因检测在实际应用中具有更高的临床价值。
4 结语与展望
忽略患者遗传信息的传统药物治疗通常会导致不同的药物反应和预期以外的治疗效果,遗传变异在一定程度上解释了药物反应的个体差异。目前,PharmGKB 收录了4 324 条药物基因数据,涉及900多个基因,主要与694 种药物的疗效、剂量和ADR相关。与ADR 相关的基因数据信息共1 544 条,其中被CPIC 等指南收录与ADR 相关的基因有13 个,药物有23 种。
发生ADR 时必须考虑以下几个关键问题:首先,需要充分了解疾病的致病因素,包括遗传和环境因素以及它们的相互作用;其次,充分考虑决定药物反应的基因和非基因因素,验证基因突变与药物反应的相关性,从而使药物疗效最大化。最后,充分明确患者的疾病诊断和种族/民族信息以避免其他遗传因素的干扰,确保基因多态性指导用药的准确性。
随着药物基因组学研究技术的不断发展,识别药物相关基因多态性特别是罕见基因变异会变得更加容易。药物基因组学正处于临床应用的早期阶段,拥有光明的前景,利用药物基因组学信息预测药物反应的遗传多态性,更加合理地设计个体化用药方案,规避ADR 风险,在成本效益的基础上实现最佳的药物治疗效果是临床长期追求的奋斗目标。
