GCDH 基因新变异致戊二酸血症Ⅰ型的遗传学分析及产前诊断
2022-11-28董兴盛王德刚李志明熊怡满婷婷
董兴盛 王德刚 李志明 熊怡 满婷婷
戊二酸血症Ⅰ型(GAⅠ)是一种常染色体隐 性遗传的代谢性疾病,由于戊二酰辅酶A 脱氢酶(GCDH)基因缺陷使GCDH 活性降低甚至缺失,导致戊二酸、3- 羟基戊二酸在体内蓄积,引起纹状体等神经核团损害,导致神经退行性病变[1]。本研究对GAⅠ家系中的先证者进行全外显子组测序及核心成员突变位点Sanger 测序验证,经分析确定变异为致病性,通过产前羊水DNA 的GCDH 基因分析及遗传咨询,提供优生优育指导,现报道如下。
对象与方法
一、一个GAⅠ家系先证者的临床资料收集
收集一个GAⅠ家系先证者的病史和体格检查资料。本研究经中山市博爱医院医学伦理委员会审核批准(KY-2019-010-37),先证者父母已签署知情同意书。
二、基因测序分析
1. 先证者外周血基因检测
采用血液DNA 纯化试剂盒(QIAgen,德国)提取先证者及家系核心成员的外周血,具体操作按试剂盒说明书进行。利用Covaris LE220 超声波仪(Massachusetts,美国)将待测基因组DNA打断成小片段DNA,进行DNA 文库构建。利用定制的基因片段捕获探针( Roche NimbleGen,Madison,美国)进行外显子组捕获,再由高通量测序仪BGISEQ-500(华大基因,中国)完成测序。人类基因组参考序列版本选择GRCh37。变异位点Sanger 测序验证:采用 Primer5 软件设计2对引物,扩增GCDH 基因的2 个位点。引物序列:GCDH-F1 为5′-GGCTAAGTGTAAGGACCTCTGG-3′,GCDH-R1 为5′-CACCTTCGTTGCGATTGG-3′;GCDH-F2 为5′-TGACCGTCTCGCTCATCCC-3′,GCDH-R2 为5′-CCGTTGACTCAGCCCACA-3′。使用PTC-200PCR 仪(Bio-Rad,美国)进行PCR,反应条件为96 ℃预变性5 min,96 ℃变性20 s,59.8/58.4 ℃退火30 s,72 ℃延伸30 s,38 个循环;最终72 ℃延伸5 min,4 ℃保存。PCR 扩增产物通过ABI3730 全自动测序仪(美国,ABI 公司)进行正反双向测序。根据美国医学遗传学与基因组学学会(ACMG)发布的遗传变异分类标准与指南对筛选出来的变异进行遗传致病性分析[2]。
2. 产前诊断
对先证者孕18 周母亲行羊膜穿刺术抽取羊水,用前述方法提取胎儿羊水DNA,并使用Sanger 测序方法对胎儿行GCDH 基因测序分析。排除母体污染标准:胎儿各位点等位基因荧光峰分别来自父母,胎儿各位点无来自母亲的第2 个荧光检测峰。
三、文献检索
选择“戊二酸血症Ⅰ型/ Glutarate disease typeⅠ”“ GCDH”及“ 产前诊断/ Prenatal diagnosis”为检索词,对以下数据库截至 2022 年6 月收录的论文进行检索:PubMed、CNKI、万方数据知识服务平台、维普中文科技期刊数据库,收集并分析检索到的GAⅠ产前诊断文献资料。
结 果
一、一个GAⅠ家系先证者的临床资料
本研究家系来自中国福建省。2021 年3 月,先证者母亲孕17 周,携先证者来本院产前诊断中心就诊咨询,其孕期常规产检结果均正常,寻求遗传学咨询以避免再次生育患病后代。患儿(先证者)女,1 岁7 个月。6 月龄时,不能抬头,可以翻身,可以抓物。7 月龄时,运动发育倒退,不能翻身,不能抓物。8 月龄时,尿有机酸分析提示戊二酸升高(66.8 µmol/L,参考值范围0.5~5.9 µmol/L);血酰基肉碱谱检测提示戊二酰肉碱升高(0.75 µmol/L,参考值范围0.05~0.40 µmol/L),游离肉碱降低(5.65 µmol/ L,参考值范围10.00~58.00 µmol/L);头颅MRI 发现双侧额颞叶脑白质低髓鞘化,双侧豆状核(苍白球为主)对称性异常增高信号。现先证者19 月龄,不会叫“爸、妈”,抬头不稳,不能翻身,不能独坐,不能爬行,不能站立,不能抓物。先证者双亲均体健,否认近亲结婚,否认遗传病家族史。先证者体格检查:体质量9.5 kg,身高77 cm,头围47 cm。颈软无抵抗,竖头不稳。心、肺听诊无异常,腹平软,肝、脾不大,四肢肌张力稍低。
二、先证者的全外显子组测序结果
家系中的先证者通过全外显子组测序,检测到GCDH(NM_000159.3)基因位于4 号外显子的杂合变异c.206_207delAC(p.Thr70Leufs*117)和位于9 号外显子的杂合变异c.892G>A(p.Ala298Thr)。见表1。

表1 先证者全外显子组测序结果
三、家系核心成员Sanger 测序验证结果
先证者基因Sanger 测序结果和全外显子组测序结果一致。先证者父亲检出GCDH:c.206_207delAC(p.Thr70Leufs*117)杂合变异;先证者母亲检出GCDH:c.892G>A(p.Ala298Thr)杂合变异。见图1。

图1 先证者及其父母的 GCDH 基因测序结果
四、致病性分析
先证者检出GCDH 基因的2 个复合杂合变异(分别遗传自父亲、母亲):①GCDH 基因(NM_000159.3)c.206_207delAC,第4 号外显子区的第206~207 位碱基GA 缺失,导致其蛋白质第70 位的苏氨酸变成亮氨酸,并产生移码突变,翻译117 个氨基酸后终止(p.Thr70Leufs*117)。该变异在gnomAD、ExAC 及1000G 数据库中未见收录。该移码预测可引起转录子降解,可使用PSV1 证据。根据2015 年ACMG 发布的序列变异解读指南,变异证据包括PM2、PSV1、PP4,该变异判读为致病变异。②GCDH 基因(NM_000159.3)c.892G>A,位于第9 号外显子的错义变异,导致蛋白质翻译在第298 位的丙氨酸变成苏氨酸。该变异在gnomAD数据库中东亚人群中等位基因频率为0.0001,属于隐性遗传病中极低频位点。根据2015 年ACMG发布的序列变异解读指南,变异证据包括PM1、PM2、PM3-strong 及PP4-moderate,该变异判读为致病变异。见表2。

表2 GCDH 基因c.206_207delAC 及c.892G>A 变异致病性分析
五、产前诊断结果
针对GCDH 基因的2 个位点进行Sanger 测序,胎儿羊水DNA 未测出母体细胞污染,产前诊断结果为胎儿的GCDH 基因变异的复合杂合变异c.206_207delAC 和c.892G>A,见图2。经遗传咨询,胎儿父母要求终止妊娠。
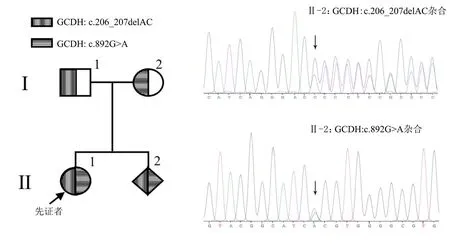
图2 家系图及胎儿GCDH 基因测序结果
六、文献检索结果
文献检索收集到GCDH 基因测序分析产前诊断GAⅠ的文献5 篇,包括9 例GAⅠ家系。其中4例家系中5 例胎儿通过GCDH 基因测序分析诊断为GAⅠ, 4 例GAⅠ胎儿家属选择终止妊娠,仅有1例胎儿家属要求继续妊娠并足月分娩。见表3。

表3 GAⅠ产前诊断的文献资料总结
讨 论
GAⅠ是一种因GCDH 基因变异导致的罕见常染色体隐性遗传的代谢性疾病。该病在全球发病率约为1∶110 000[8-9]。GCDH 基因定位于染色体19p13.2,约7 kb,包含11 个外显子,编码438 个氨基酸组成的前体蛋白,其中N 端44 个氨基酸是线粒体定位信号,在进入线粒体后被切除[5]。4 个单体构成的同源四聚体即GCDH,是一种同源四聚体黄素蛋白,属于酰基CoA 脱氢酶家族,负责催化戊二酰CoA 脱氢和脱羧成为巴豆酰辅酶A 和CO2[10]。GCDH 单体的二级结构分成3 个结构域:N端α-螺旋(R 45 ~ Q 167)、C 端α-螺旋( S 282 ~K 438)及中间的β-片层(L168 ~S 281),C 端ɑ-螺旋结构域是主要的催化活性中心[7]。GCDH 基因发生变异时,酶的正常结构受到影响,导致戊二酸和3-羟基戊二酸在体内蓄积。戊二酸和3-羟基戊二酸与兴奋性精神递质谷氨酸结构相似,导致谷氨酸受体过度激活,抑制γ-氨基丁酸(GABA)的合成,使抑制性神经递质减少,从而对神经元造成损伤[11]。目前人类基因组突变数据库(HGMD)已报道人类GCDH 基因致病变异290 种,其中错义突变是最常见的类型。不同的种族群体存在不同的GCDH 基因变异热点,c.1244-2A>C是中国人群中较为常见的热点变异,c.1296C>T在美国人群较为常见,c.1204C > T 在欧洲人群中最为常见,c.914C>T 在日本人群中的携带率高达12.1%[12-17]。
本例家系患者检出了GCDH 基因的c.206_207delAC(p.Thr70Leufs*117)及c.892G>A(p.Ala298Thr)变异。其中c.206_207delAC 变异为首次报道,在GCDH 基因c.206_207 位置的缺失导致移码,GCDH 蛋白第70 位的苏氨酸变成亮氨酸,之后翻译117 个氨基酸即终止(p.Thr70Leufs*117)。该移码突变导致编码肽链减少251 个氨基酸残基,形成截断蛋白。有研究显示,GCDH 基因的移码突变可导致基因功能丧失,GCDH 活性完全缺失的GAⅠ患者出现更严重的表型[18]。c.892G>A 变异是已有文献报道的已知变异,在多例GAⅠ患者中观察到了这种变异[19-22]。该位点在不同物种中高度保守,位于GCDH 蛋白单体的C 端α-螺旋结构域。该结构域是GCDH 主要的催化活性中心,变异会影响该酶的催化活性。Christensen 等[20]发现c.892G>A 变异纯合子的GAⅠ患者的成纤维细胞中GCDH 活性明显降低,仅为正常活性的5%~10%。
GAⅠ的临床表现多样,患儿常于6~18 月龄时发病,主要的临床特征为大头畸形、肌张力异常、进行性运动障碍等,这是由于GCDH 活性降低或缺失导致戊二酸、3 -羟-戊二酸、戊烯二酸及戊二酰肉碱等有机酸在体内升高[23]。由于患者的表型谱变异度大,即使具有相同基因型的家系患者之间的表型可能有很大差异。大头畸形是GAⅠ的一个主要临床特征,也是70%GAⅠ患儿在新生儿期出现的第一个体征,所以对于大头畸形的新生儿应注意排除GAⅠ[24]。本例先证者新生儿期头围正常,故未行GAⅠ相关的筛查和基因诊断。根据ACMG 指南,GAⅠ的诊断必须基于GCDH 活性降低和(或)检测出2 个GCDH 等位基因的致病变异[9]。先证者的GCDH 基因结果符合诊断,临床表现与GAⅠ一致,如肌张力减退、运动发育迟缓、体液中戊二酸和戊二酰肉碱浓度升高、头颅MRI提示脑白质和苍白球的改变。结合GCDH 基因的结果和临床表现,先证者确诊GAⅠ。
GAⅠ可通过检测羊水中戊二酸水平或羊膜细胞中戊二酰辅酶A 脱氢酶的活性进行产前诊断,但临床存在戊二酸盐排泄正常和有显著残留活性的GAⅠ患者,因此仅依靠生化指标难以得到精确的产前诊断结果[3]。2000 年Busquets 等[3]首次通过GCDH 基因测序分析在产前诊断GAⅠ。本研究总结了通过GCDH 基因测序分析进行GAⅠ产前诊断的文献,5 例胎儿通过GCDH 基因测序分析产前诊断为GAⅠ,其中4 例胎儿父母选择终止妊娠;仅有1 例胎儿家属要求继续妊娠,该例孕晚期时胎儿超声检查结果显示大头畸形、额颞叶萎缩和大脑外侧裂增宽,足月剖宫产分娩,出生后接受谷氨酸特殊配方奶粉治疗,由儿科内分泌科医师定期监测随访[4]。本研究的先证者母亲再生育时,也通过GCDH 基因测序分析进行产前诊断,结果明确胎儿为GCDH 基因的复合杂合变异(c.206_207delAC 和c.892G>A),诊断胎儿罹患GAⅠ。妊娠20 周时胎儿超声检查未见明显异常。经遗传咨询,胎儿父母要求终止妊娠,妊娠22 周引产。家属拒绝进行尸检,故未对引产胎儿进行尸检。
综上所述,分子遗传学检测是GAⅠ的有效确诊方法。本研究报道1 例GAⅠ家系中GCDH 基因复合杂合变异,并发现1 个新的移码突变,丰富了HGMD,有助于更好地了解该疾病的分子病理学,并为该家系的遗传咨询及产前诊断提供了遗传学依据,从而有效地避免该家系再次生育缺陷儿。
