生物类似药Ⅲ期临床比对研究中的关键问题分析
2021-08-11韩静静李文倩徐润泽杨劲
韩静静,李文倩,徐润泽,杨劲
(中国药科大学药物代谢动力学重点实验室,江苏 南京 211198)
2019年全球十大畅销药有7个为生物药,生物药如此畅销的原因除了其具有巨大的临床价值外,更重要一点的是没有其“拷贝版”进入市场产生价格竞争,生物药的价格居高不下,给整个社会带来巨大的经济负担。在生物药的专利到期后,非原研公司可以对生物药进行“拷贝”,为此监管机构引入生物类似药的概念,即结构、生物活性、安全性和有效性等与已批准的参考生物药高度相似的生物制剂[1-4]。由于生物药的结构复杂性,生物类似药远比小分子仿制药评估方法复杂。欧洲药品管理局(EMA)与美国食品和药品管理局(FDA)对于生物类似药评价的基本原则是通过逐步比对的方法,即在药学比对的基础上,分别在非临床、临床药理、临床有效性和安全性研究中进行比对,建立生物类似药与生物原研药之间的相似性[2,5-7]。
生物类似药在临床研发阶段一般分为Ⅰ期和Ⅲ期,在Ⅰ期临床研究阶段,主要将候选药和对照药进行药代动力学(PK)比对研究和药效学(PD)比对研究,而在Ⅲ期临床研究阶段,则主要围绕与对照药的有效性和安全性的比对研究。与新药审批路径相比,生物类似药审批途径的一大价值在于可以选择原研药的众多适应证中的一个或几个适应证进行临床比对研究后再将相似的结论外推到其余多个适应证,无需再进行在其余适应证上的临床有效性试验,能较大程度地降低对临床试验的成本投入。这就需要申办方在Ⅲ期临床比对中对适应证、临床终点和研究周期的选择及等效界值的确定等关键点进行充分考虑。如英夫利昔单抗(infliximab)获批了多个适应证,申办方该选择哪一适应证进行比对,什么是敏感的临床模型,什么情况下可以外推至其他适应证?虽然等效/非劣效标准是刚性要求,但在审评实践中存在适当被放宽的特殊情况[8],监管机构如何科学考量?另外,非格司亭的生物类似药依据PK/PD比对数据豁免了Ⅲ期临床比对研究,其他药物是否可以借鉴?研究显示,在未来几年,随着多种原研药专利到期,批准的生物类似药的数量将大幅增长[9-10]。本文对EMA与FDA已上市的生物类似药的Ⅲ期临床研究的案例进行梳理,概述了确定敏感临床模型的关键要素、Ⅲ期临床比对研究的结果在审评实践中的科学考量以及Ⅲ期临床比对研究的豁免等问题,以期为中国研发更复杂的生物类似药的Ⅲ期临床研究策略提供参考。
1 Ⅲ期临床比对研究中的敏感模型
与新药的Ⅲ期临床研究需要证明候选药优效于对照药不同,在生物类似物的开发中,Ⅲ期临床比对研究是在预先设定边界的基础上,在特定的患者中进行候选生物类似药与参照药的疗效比对,确认候选生物类似药与生物原研药之间无临床意义的差异。然而,通过对EMA批准的曲妥珠单抗(trastuzumab)生物类似药的Ⅲ期临床比对(见表1)案例分析可见,同一原研药的多个生物类似药在临床开发中遵循的临床研究策略并不同,如在患者群体、临床终点选择和临床试验设计方面均存在差异[11]。针对以上问题,监管机构提出了敏感模型的概念,涉及适应证、临床终点以及研究周期等关键因素的敏感性。
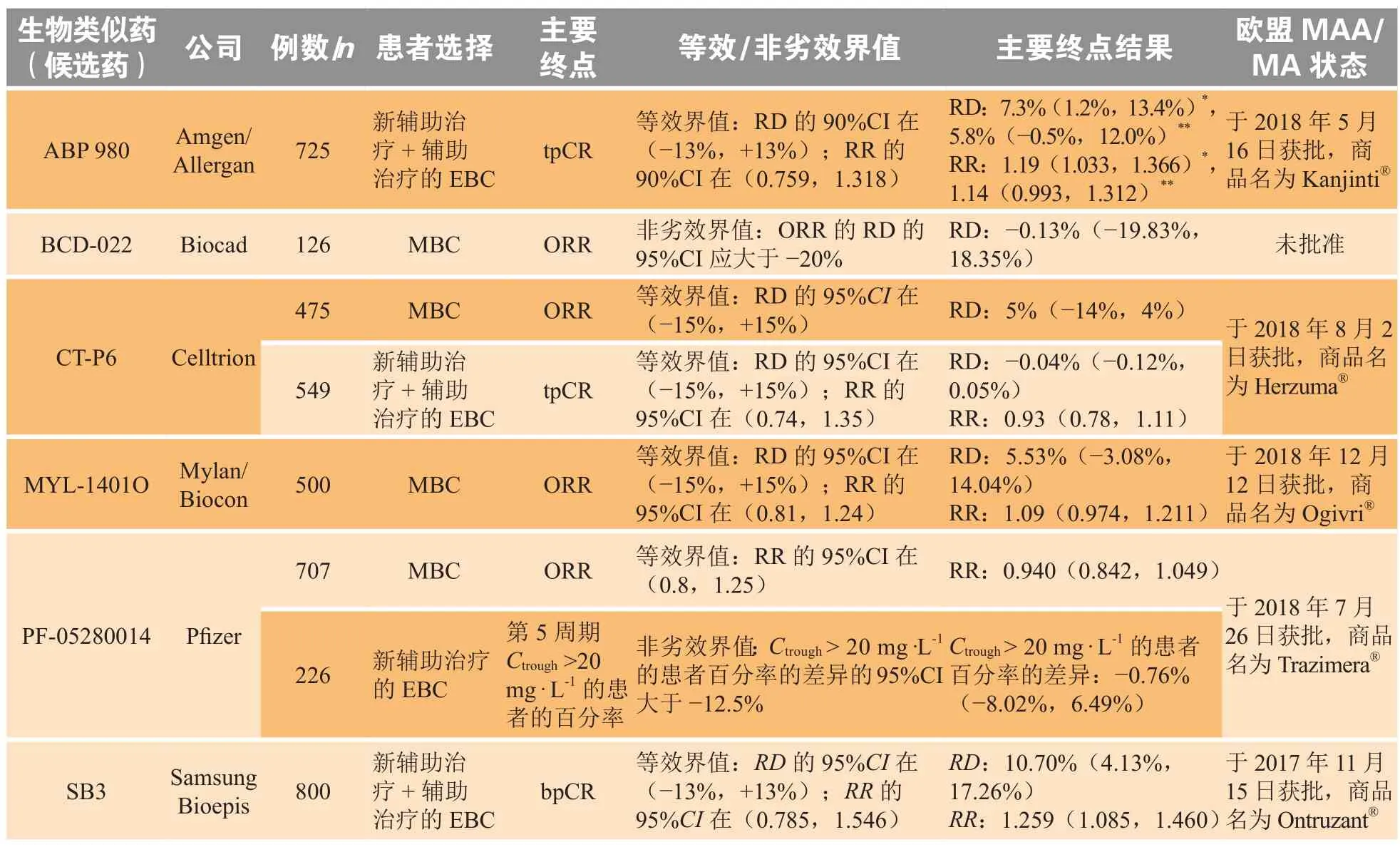
表1 曲妥珠单抗生物类似药(候选药物)的Ⅲ期试验参数和主要终点结果Table 1 PhaseⅢtrial parameters and primary endpoint results for trastuzumab biosimilars (candidates)
1.1 适应证与患者亚组人群的选择
生物类似药的Ⅲ期临床比对试验需要在患者身上观察疗效的可比性,但是当原研药已经获批多个适应证时,单抗类药物(见表2)获批了多个适应证,必然需要考虑从中选择一个适应证进行临床研究后,再通过适应证外推的方法获准所有适应证[17]。监管机构如EMA和FDA通常建议疗效比对试验应该在敏感且同质性人群中进行,以检测是否存在临床意义的差异[18]。这个考虑要点包含了2个方面:选择敏感的适应证和敏感同质的患者亚组人群。

表2 美国专利到期且具有重大临床价值的单抗类药物的适应证批准情况Table 2 Approval of indications of monoclonal antibodies with US patent expiry and yet still significant clinical value
敏感适应证是指假设同一给药剂量,对于2种不同的适应证(见图1),生物类似药候选药与参照药的微小差异在适应证2中的响应差异大于在适应证1中的响应差异,则适应证2比适应证1的敏感,更易于检测两药之间的差异。有学者认为适应证的敏感性与原研药在该适应证人群中所表现的临床效应量的大小有关,效应量越大,候选生物类似药与原研药在临床意义上的差异越容易凸显出来[18]。例如曲妥珠单抗的适应证有早期乳腺癌(EBC)、转移性乳腺癌(MBC)和转移性胃癌,EMA和FDA批准的所有5个曲妥珠单抗的生物类似药(见表1)的Ⅲ期临床比对多是在MBC和EBC患者中进行研究,而没有在人体表皮生长因子受体-2(HER2)阳性的胃癌患者中进行。因为与乳腺癌相比,HER2阳性转移性胃癌从曲妥珠单抗治疗中获益的程度较小,不易观察到差异,转移性胃癌并非开展临床有效性比对研究的敏感适应证[19]。
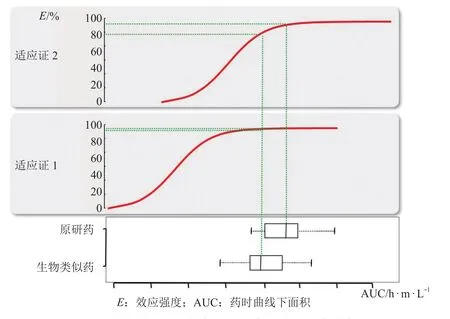
图1 生物类似药与原研药在不同适应证中的临床响应差异Figure 1 Differences in clinical response between the biosimilar and the original drug in different indications at the same exposure level
患者入组时应充分考虑亚组人群的选择,例如在克罗恩病患者细胞中,如果氨基酸残基158位的基因型为V/V和V/F,英夫利昔单抗生物类似药与FCGRⅢa(FC gama receptorⅢa)的结合亲和力低于原研药,但在F/F基因型中2种药物结合情况相似[20]。这个结果表明,比对结果可能受患者基因型的影响,所以在临床比对中需要充分评估敏感人群。因此,虽然监管机构没有指定临床研究应该选用哪一种适应证进行研究,但若选择的人群不够敏感,很可能会影响试验的有效性结果,或者需要更大样本量来保证临床相似性,不仅增加了生物类似药临床试验的成本,还不利于其后期适应证的科学外推。
1.2 临床终点与研究周期的选择
生物类似药与对照药在进行Ⅲ期临床比对时,应选择足以检测两药之间临床相关差异的最敏感的临床终点,可以理解为生物类似药和对照药在这个临床终点上的响应差值更易于被检测到。临床终点的选择通常应该是参考产品临床试验中使用的一个或多个终点,如果存在合适的替代指标能够反映药品的临床疗效,可以考虑将衡量短期内活性的替代临床终点作为主要终点[5]。
曲妥珠单抗原研药在临床研究中使用的终点为 无 病 生 存 期(disease-free survival,DFS),EMA与FDA批准的曲妥珠单抗生物类似药的临床比对研究中,使用的主要终点为MBC患者的客观缓解率(ORR)和EBC患者的病理完全缓解率(pathological complete response,pCR)。研究证实,pCR与患者的长期生存率相关,且其治疗观察时间较短,可以有效地缩短研发周期,是一个敏感的临床终点[21]。
关于研究周期的问题,应同时考虑有效性和安全性的评估时间。对于慢性疾病,如自身免疫性疾病,临床反应通常很慢,Ⅲ期临床比对研究应有足够长的研究周期以观察生物类似药的有效性、安全性以及免疫原性特征。在目前的慢性疾病临床研究中,研究者们一般倾向于评估与长期疗效相关的临床终点,但对于证明生物相似性的比对试验来说,长时间试验的一个可能的缺点是纵向偏倚,如患者退出的情况,这可能会影响疗效比对。这种情况下如果存在疗效评估时间较短的替代终点,就能减小纵向偏倚,使得该临床比对试验对两药潜在差异的检测更加敏感。
综上,针对众多可选择的临床终点,申办方应选择敏感且与临床终点相关的替代指标,在考虑安全性观察周期的同时,寻找使两药疗效差异表现最大的时间点,以便在检测两药潜在差异的同时节约试验成本。
1.3 剂量选择
监管机构建议候选药的给药剂量应在参照药批准的用量范围内。在患者人群中,一般应采用最低治疗剂量评估原研药和生物类似药的单次给药的疗效比对。剂量的敏感性与该剂量在原研药的剂量-反应曲线上的位置有关:当剂量足够高时,剂量-反应曲线达到平台期,如图2中的剂量1,此时,候选药与原研药的差异几乎不能被检测出来;但是当剂量相对低时,如剂量2,此时,候选药与原研药的差异在临床响应上凸显出来,所以剂量2相对于剂量1更敏感(见图2)。一般来说,剂量-反应曲线上最陡峭的位置最敏感,但是,目前使用的生物制品很多是饱和给药的,所以一般是在暴露量-效应曲线平台期上,此时候选药与原研药的临床意义的差异不易被检测出来(见图2)[22]。

图2 在同一适应证人群中不同给药剂量下原研药与候选药的临床响应差异Figure 2 Clinical response difference between the original drug and the candidate under different doses in the same population
1.4 等效界值
新药的关键性临床试验为优效试验,其目的是证明试验组疗效优于对照组,但这不适用于生物类似药的研发。从表1中可以看出Ⅲ期临床研究中一般选择等效性试验,其中一些非劣效试验并没有作为证明相似的临床数据来使用。关于生物类似药的Ⅲ期临床比对研究,FDA建议一般采用等效性试验设计,并且在试验进行之前预设等效界值[3]。等效界值的大小影响着试验所需样本量的大小,等效界值越大,所需样本量越小。但FDA也指出,在一些特殊情况如研究中所使用的剂量已经接近剂量-反应曲线平台且几乎不可能有剂量相关的影响时,可以使用上边界稍大的非对称区间,甚至使用非劣效试验设计来证明两药的相似性[3]。如英夫利昔单抗的生物类似药Inflectra和Renflexis的临床比对的等效界值为(-12%,12%),而另外2个生物类似药Lxifi和Avsola的临床有效性的等效界值设定为(-12%,15%)。一般来说,使用上边界稍大的非对称区间比使用对称区间所需样本量更小,也意味着这个试验更容易得到等效的结果且经济成本较低。
综上所述,生物类似药的Ⅲ期临床比对研究在整个研发过程中消耗巨大的经济成本和时间成本,对于临床试验而言,良好的临床策略能够提高试验的成功率并降低成本。同一原研药产品的生物类似药的临床比对中的适应证选择、临床终点选择、等效界值的大小等因素均会影响最终预估的样本例数,需要申办方对这些复杂的要素综合权衡,以选择最优的临床研发策略。
2 Ⅲ期临床比对研究的结果在审评实践中的科学考量
生物相似性是由完整证据的总和来确定,进行Ⅲ期临床比对研究是为了解决前期比对中存在的剩余不确定性。研究发现,在所有已经批准上市的生物类似药中,药学比对和在人体中的PK比对结果是必须相似的[23],但值得注意的是,存在生物类似药Ⅲ期临床比对中的疗效比对不相似但仍被批准的案例[11]。后文以案例分析的形式对曲妥珠单抗的2个生物类似药ABP980、SB3进行剖析,探讨如何科学考量Ⅲ期临床比对研究的结果。
曲妥珠单抗(商品名:郝赛汀)是一种抗HER2的单克隆抗体,适应证为MBC、早期乳腺癌和转移性胃癌。随着原研药的专利到期,曲妥珠单抗的生物类似药陆续被开发上市,但Kanjinti®(ABP980)和Ontruzant®(SB3)的疗效比对结果并不在预设等效界值之内,不能证明疗效等效[11]。最后监管机构与申请人讨论后,认为该候选药物与参照药相似,批准其上市。临床试验希望得到的结果是疗效相似,试验疗效不等效却被批准,值得仔细分析其过程。
2.1 生物类似药ABP980案例分析
ABP980关键临床研究是一项大型的、国际化、多中心试验,所选研究人群为新辅助治疗+辅助治疗的EBC患者,主要终点为患者的总病理完全缓解率(total pathological complete response,tpCR),评估其风险差异(risk difference,RD)和风险比(risk ratio,RR)与参照药的RD和RR的等效性,预设界值为RD的90%CI为(-13%,13%),RR的90%CI为(0.759,1.318)[24]。最初申请人选择本地研究者评估的方法,RD的90%CI为7.3%(1.2%,13.4%),下限在预设界值内,上限超过了预设界值,RR的90%CI为1.19(1.033,1.366),下限在预设界值内,上限超过了预设界值,同时安全性和免疫原性结果相似,不良事件的频率、类型和严重程度(包括心脏事件)与参照药之间无差异。但是,在一项基于肿瘤样本的中心阅片的敏感性分析中,2种药的RD和RR的90%CI分别为5.8%(-0.5%,12.0%)和1.142(0.993,1.312),均在等效界值内[25]。根据ABP980的欧盟公众评估报告(EPAR),欧洲人用药品委员会(CHMP)在本地研究者评估的基础上考虑了tpCR的RD和RR的95%CI,同样,其95%CI的上限也均超过了预先规定的界限[25]。针对这种情况,CHMP认为这可能是由于所使用批次的参照药的抗体依赖的细胞介导的细胞毒性作用(antibody-dependent cell-mediated cytotoxicity,ADCC)功能的下降导致。所以,CHMP认为,观察到的疗效结果差异不具有临床意义,并得出结论:ABP980和曲妥珠单抗参考产品在HER2阳性EBC患者中的安全性、有效性和临床结果无差异[8]。
2.2 生物类似药SB3案例分析
SB3关键性临床研究中的研究人群为新辅助治疗+辅助治疗的EBC患者,主要终点为乳房病理学完全缓解率(breast pathological complete response,bpCR),评估其RD和RR与原研药的等效性,预设界值为RD的95%CI为(-13%,13%),RR的95%CI为(0.785,1.546)[26]。该研究的比对结果为,bpCR的RD的95%CI为10.70%(4.13%,17.26%),其上限也超过了等效界值,只能证明非劣效,其RR的95%CI为1.259(1.085,1.460),落在了预先设定的等效界值内。针对这种情况,CHMP认为,bpCR的上限略超过等效界值的原因也是由于所使用批次参照药的ADCC功能的下降所导致。ADCC为曲妥珠单抗的作用机制之一,参照药的ADCC功能下降,可能导致临床终点的响应变化,结合其他部分的比对证据,CHMP认为观察到的差异不能证明其具有临床意义的差异,不能否定该生物类似药与原研药相似的结论[26]。
综上所述,通过对曲妥珠单抗的生物类似药的案例分析可知,在特殊情况下,当其他部分的证据能充分证明生物相似且能保证安全性的情况下,对于生物类似药的临床疗效可能优于原研药的不确定性的存在,监管机构是可以接受的,但是如果存在劣效的不确定性是不能接受的[4]。另有研究亦认为,生物相似的结论不能根据某一步骤的比对结果来判定,而应该把所有比对证据放在一起进行整体评估[27]。因此,Ⅲ期临床比对的证据在整个证据链中起验证作用,用于解决药学比对、PK/PD比对等步骤后存在的剩余不确定性,如果Ⅲ期临床比对观测的微小差异不会产生临床意义的差异,则不能用于否定候选药与原研药相似的结论。
3 Ⅲ期临床比对研究的豁免
近年来,EMA认为部分产品类别(胰岛素、低分子肝素和非格司亭)没有必要进行专门的有效性比对试验,其相似性的关键证据可以来自于生物类似药和原研药的物理化学、功能特征、PK和PD比对结果[28-30]。此外,如果公认的具有低免疫原性风险的生物类似药的杂质和赋形剂的性质不会有很大的影响,则可能豁免生物类似药的安全性和(或)免疫原性研究[28-30]。表3为EMA对各种类别的生物类似药所需进行的临床试验的建议[4]。后续以非格司亭为案例解释Ⅲ期临床比对研究是如何被豁免的。

表3 对不同产品类别生物类似药的临床研究建议Table 3 Clinical study recommendation for the biosimilar of different product categories
非格司亭又称重组人粒细胞集落刺激因子,临床上用于中性粒细胞减少症。由于非格司亭结构、理化特性和生物活性均可以很好地表征,且有与临床疗效相关的PD指标,即嗜中性细胞绝对计数(ANC),EMA关于生物相似的重组粒细胞集落因子的指南修订草案[30]指出,没有必要对其生物类似药进行专门的比较疗效试验。FDA虽然没有针对粒细胞集落因子的个药指南,但是从FDA对非格司亭生物类似药产品Nivestym的审评上也能看出FDA对于该药是否需要临床疗效比对的态度。在Nivestym的申报过程中,申办方一共递交了3份临床试验报告,包括一项Ⅰ期单剂量的PK/PD研究、一项Ⅰ期多剂量的PD研究和一项用于比较免疫原性的多剂量研究[31]。Nivestym的研发过程中没有进行Ⅲ期临床疗效比对试验,其有效性数据是基于PK/PD的比对结果,安全性评估是基于在健康人体中的Ⅰ期试验。因此,有研究认为合适的PD指标通常比临床终点具有更好的敏感性,更小的变异以及较短的响应时间,利用PK/PD比对这种简单方法来代替复杂的Ⅲ期临床疗效比对也是未来药品监管科学发展的重要方向[32]。
目前,能够豁免临床有效性比对试验的仅限于一些相对分子质量小的生物类似药,不过随着单抗类药物的PD指标的发现[33],或许将来能利用PK/PD比对这一数据豁免更多的生物类似药的Ⅲ期临床比对试验,这将大大降低生物类似药的研发成本,减轻社会医疗支付压力。除了EMA和FDA的政策外,英国药品与医疗保健产品监管机构(MHRA)刚公布了一项生物类似药指南草案,通过取消大多数情况下对生物类似药进行Ⅲ期临床比对疗效试验的需求,来减少英国对生物类似药申报的临床试验数据的要求[34],这一做法或许有些激进,但也表明了MHRA渴望尽快降低生物类似药临床研发成本的态度。
4 结语与展望
由于生物药的结构复杂性,申办方在生物类似药研发中应逐步分析,建立候选药和对照药之间的相似性,当前期比对中存在剩余不确定性时,需进行一项或多项Ⅲ期临床比对研究来保证生物类似药与原研药无临床意义的差异。在临床比对中会面临如何确定敏感模型的问题,需要综合考虑各个关键因素的敏感性,其本质是把生物类似药与原研药在临床上的差异最大化地表现出来。另外,敏感模型的选择除了影响试验的成功率以及研发成本外,还对生物类似药的适应证外推十分重要,在敏感模型中的比对数据是适应证外推的科学证据之一,因此申办方需进行充分评估和比较,并积极与监管机构紧密互动,以选择最佳临床研发策略。
目前,利用PK/PD比对来代替Ⅲ期临床比对研究的均是一些相对分子质量较小的生物药,对于复杂的单抗类药物,与临床疗效相关的PD指标的发现对该单抗类的生物类似药的Ⅲ期临床比对研究的豁免极其关键,因为利用PK/PD比对来表征疗效相似可以简化生物类似药的临床开发过程,提高研发效率,同时也最大限度减轻生物类似药的临床开发负担,加快生物类似药上市。
在未来几年,随着生物原研药(尤其是在肿瘤领域)的专利到期,我国批准的生物类似药的数量将大幅增长,如何更快、更高效地开发有效、安全可控的生物类似药是监管者与申办方的共同目标和挑战。国家药品监督管理局药品审评中心近期发布了《生物类似药物相似性评价和适应证外推技术指导原则》的征求意见稿,目的是推动国内生物类似药的发展。期待着我国药品监管科学研究的快速发展,及时制定相关生物类似药个药指南,为生物类似药研发提供科学的建议。
