基于Context建模熵编码的基因组序列应用
2021-07-15陈慧
陈慧

摘 要:该文通过将生物学特征和生物学含义引入DNA序列数据的压缩处理中, 提出了基于生物信息学特征的基因组序列的Context建模熵编码技术,拟结合基因组序列特点,研究针对基因组序列的Context建模熵编码技术。在算法中DNA序列根据组成部分生物学含义的不同切分重组为4个集合:编码序列CDS集合、内含子序列集合、RNA序列集合以及剩余序列的集合。根据各集合中序列的具体生物学特征分别进行预处理, 并通过熵编码算法进行压缩。实验结果表明,该算法在基准测试序列上的压缩性能优于原有的DNA序列压缩方法,特别是对于生物信息学特征清晰的长序列,算法能够在较短的时间内获得较高的压缩率。
关键词:基因组序列 Context建模 熵编码 集合
中图分类号:G64 文献标识码:A文章编号:1672-3791(2021)03(c)-0025-03
Application of Entropy Coding Genome Sequence Based on Context Modeling
CHEN Hui
(Dianchi College of Yunnan University, Kunming, Yunnan Province, 650228 China)
Abstract: In this paper, by introducing biological characteristics and biological meaning into the compression processing of DNA sequence data, Context modeling entropy coding technology of genome sequence based on bioinformatics features was proposed. It is intended to combine the characteristics of genome sequence to study the context modeling entropy coding technology of genome sequence. In the algorithm, DNA sequences are reorganized into four sets according to the different slices of the biological meaning of the constituent parts: the CDS set of the coding sequence, the intron sequence set, the RNA sequence set and the remaining sequence set. According to the specific biological characteristics of the sequences in each set, the sequences were preprocessed and compressed by entropy coding algorithm. The experimental results show that the compression performance of the proposed algorithm is better than that of the original DNA sequence compression method, especially for long sequences with clear bioinformatics features, the algorithm can obtain a higher compression rate in a relatively short time.
Key Words: Genome sequence; Context modeling; Entropy coding; Congregation
基因组序列要保持尽可能多的遗传特性,则注定其基因组序列中要维持最低的重复片段。重复序列片段过短导致在Context建模过程中,直接选取近邻碱基来构建条件概率分布未必能够保证碱基间的相关性得以最大限度发挥。而文献[1]中指出,基因组中碱基间存在长程相关性。这就意味着,与当前碱基相关的那些碱基未必在其附近。因此,在对基因组序列进行压缩时,不能简单地按照碱基在序列中的顺序进行建模和编码。一种对基因组序列进行重新排序,使得排序后相邻碱基间的相关性得以增强,然后再对重排序后的序列进行编码以充分发挥熵编码性能。虽然重新排序有可能造成接收端由于不知道编码顺序而无法解码,但只要找到合适的方法,使得收发双方能够事先确定重新排序后的碱基顺序,则可避免上述问题的出现。而对于重排序方法,对基因组序列进行重新排序预处理,以充分利用碱基间的相关性从而提高压缩效率。
1 研究针对基因组序列的Context建模
针对基因组序列的Context建模不仅要使用传统建模方法,还要考虑使用基因组序列的生物学特征帮助建模。换言之,那些已经被生物学界确定的具有一定遗传特性的片段也应当作为条件来构建相应条件概率分布,从而对Context模型进行补充。另外,在前期研究中发现,当给定充足的微生物基因组训练序列时,不论模型阶数如何变化,得到的某些条件概率分布总是趋于均匀分布。此时,由于训练数据充足,表面上看“模型稀释”问题不存在,但其实均匀化本身也应当被看作是一種特殊的“稀释”问题。但对此问题,直接进行合并操作并不能够缓解“均匀化”,也就是说传统的Context量化方法并不适合。一种可缓解“均匀化”的思想是对计数向量进行分裂而不是合并。分裂其实是增加条件的过程,从理论上说可以降低熵值。
针对基因组序列Context建模中出现的“均匀化”问题,拟采用Context模型分裂的方法进行处理。Context模型分裂其实等价于增加条件。然而,要从低阶模型进行分裂得到高阶模型是不可行的。一种想法是在给定训练数据的前提下,先建立一个足够大阶数的模型,然后自底向上逐步合并,从而找到一个描述长度最短的编码模型。在合并过程中,模型的阶数实际上是在减少的,但如果事先给定的阶数足够大,则相对目前的建模方法,可以近似看作是一种分裂操作。在“分裂”过程中,一旦出现“均匀化”则合并停止,甚至回朔到上一级模型,从而可以避免整个Context模型的“均匀化”问题。
2 基因组序列的混合压缩算法
一方面,基于字典的压缩算法(基于Lz77的基因组压缩算法)对大量包含重复序列的物种的基因组序列具有较好的压缩效果,而单纯地使用字典类压缩算法对基因组序列进行压缩效果并不明显。但这并不意味着基于字典的压缩方法不能应用于基因组序列压缩。另一方面,Context建模熵编码技术对于非重复序列以及那些较少包含重复序列的基因组进行压缩的效果较好。因此,一种直观的想法是将两大类压缩算法进行综合。对于微生物基因组而言,应当是以Context建模熵编码为主,而使用基于字典的压缩算法对重复序列进行压缩,从而提高压缩效果。结合字典压缩算法和Context建模熵编码的微生物基因组序列压缩算法进行研究。
设Lm为计数向量Cm对应的描述长度,Lk为计数向量Ck对应的描述长度,Lmk为上述两个计数集合并后的计数集Cmk={n1(mk),…,nI(mk)}={n1(m)+n1(k),…,nI(m)+nI(k)}对应的描述长度。若nm,nk分别为Cm和Ck的总计数值,则Cmk的总计数值为nmk=nm+ nk。由此可以得到描述长度增量?Lmk的近似表达式:
(1)
从式(1)中可知,描述长度增量实际上等价于两个相对熵的平均,且满足对称性。然而,描述长度增量并不满足三角不等式。这是因为描述长度增量选取了一个共有的参考点(参考计数向量)来实现相似性的表达,即合并后的计数向量Cmk(式中计算相对熵的Cmk)由此计数向量估计得到)。这个参考点跟两个计数向量Cm和Ck是相关的,是动态的。而在物理学中,一个距离测度(相似测度)是一个相对量,而且参考点往往是静态的,例如空间坐标系中的原点(全零坐标点)。因此,在研究计数向量相似测度时同样需要考虑使用一个静态参考点。
3 仿真实验
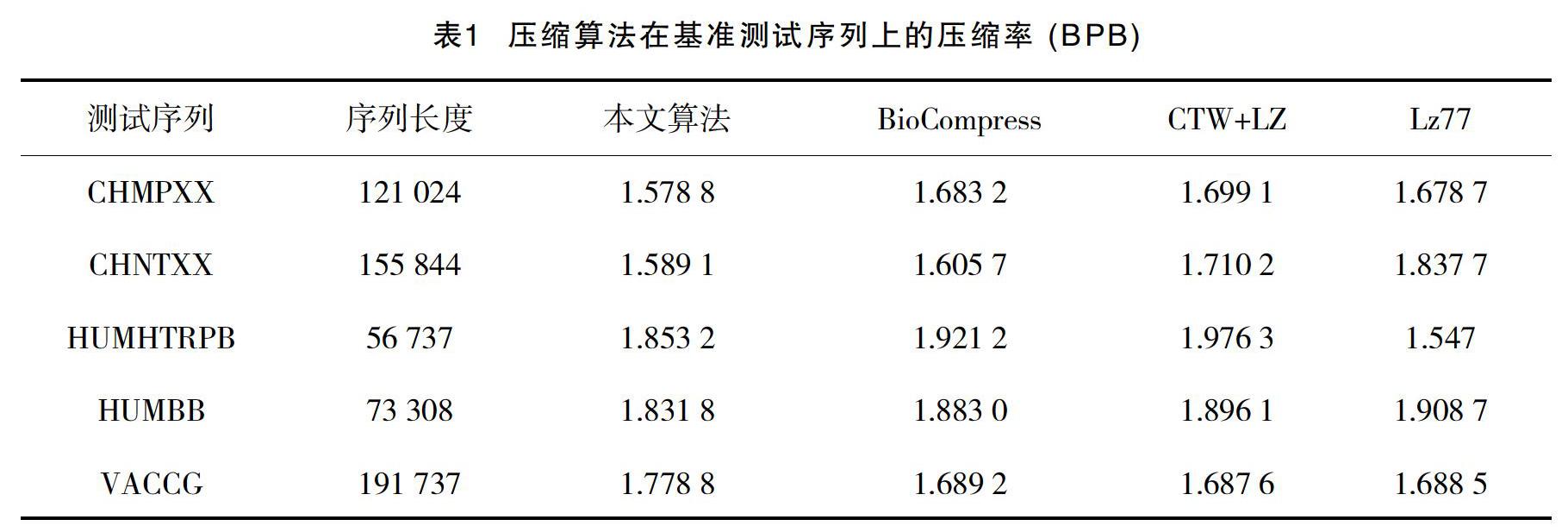
实验中使用美国GenBank数据库的DNA序列文件作为原始数据[2-3],其中包含了对序列的详细注释。在熵编码算法中, 可直接读取这些注释, 根据其生物学信息对序列进行压缩。在实验中, 将熵编码算法与典型DNA序列压缩方法:BioCompress、CTW+LZ和Lz77分别作用于5个基准测试序列[4-6]。使用压缩后序列中表示每碱基符号所需平均比特数以及熵编码算法的压缩时间作为实验结果。基准测试序列包含了不同物种不同功能的DNA数据片断, 能够有效评估压缩算法对含有不同数据特性的DNA序列的压缩能力,算法压缩率见表1。
由熵编码算法与原有DNA序列压缩算法的结果对比可以发现, 该算法在大多数基准测试序列上的压缩率要好于原有方法。特别当序列包含生物信息学特征清晰时, 算法压缩效果的提升更为明显。对于含义划分不清, 或是未包含注释信息的DNA序列数据, 依赖于生物信息学特征的熵编码算法效果并不十分理想。另外, 当DNA序列长度较大时算法性能提升更为明显,这是由于数据较长时, 其包含的重复片断也较多, 能够寻找到更多的隐含模式, 从而可有效地进行压缩编码。由结果看出,对DNA序列的生物信息学特征的序列数据进行了预处理后,产生的二进制数据流比原始序列数据更具有规律性,从而能够更为有效地进行压缩。
4 结语
该文介绍了DNA序列数据的常见生物信息学特征,通过将这些特征引入DNA序列的预处理, 提出了熵编码基因压缩算法。在算法中,含有不同生物学含义的片断被切分重组为4个集合, 通过优化序列附加信息的表示方式,算法进一步提升了压缩率。熵编码算法能够有效压缩DNA序列数据,与原有仅考虑DNA数据特点的算法相比, 使用了生物信息学特征的熵编码算法压缩性能有所提升,特别是在生物信息学特征清晰的长序列上, 其压缩结果优势更为明显。
参考文献
[1] 王灿灿.具有固定长度码字的Context自适应二进制算术码[D].云南大学,2019.
[2] 罗迪.基于最短码长的Context加权编码[D].云南大学,2015.
[3] 陈建华,王勇,张宏.基于描述长度的Context建模算法[J].电子与信息学报,2016,38(3):661-667.
[4] 罗迪,陈旻,王晴晴.基于最短码长的Context加权编码[J].计算机光盘软件与应用,2014,17(9):9-14.
[5] 陈旻,陈建华.优化Context建模及其在微生物基因组序列和图像压缩中的应用[D].云南大学,2018.
[6] 孔令超,陳建华.基于Context树建模的基因组序列无损压缩研究[C]//2020中国信息通信大会论文集.中国通信学会:人民邮电出版社电信科学编辑部,2020:5.
