Bi替代CH3NH3Pb1-xBixBr3钙钛矿实现带隙调控
2021-02-10纪登辉董润宇封顺珍张聪敏史少辉朱雪刚刘彦军李秀玲
纪登辉, 董润宇, 封顺珍*, 张聪敏, 史少辉, 李 梅, 朱雪刚, 刘彦军, 李秀玲
(1.石家庄学院 物理学院 机电学院,河北 石家庄 050035; 2.石家庄学院 图书馆,河北 石家庄 050035; 3.河北师范大学 物理学院 河北省新型薄膜材料实验室,河北 石家庄 050024)
在目前能源危机的背景下,太阳能因其取之不尽,且不产生任何污染的优势脱颖而出,成为人类可持续发展能源的选择之一[1],从19世纪末发现且成功制备第一块太阳能电池至今[2],一百多年的发展使得传统晶硅电池转化效率达到了27.6%[3].然而,目前传统硅型电池因制备繁杂成本较高,转化效率再无大幅提高而逐渐被替代.如今有机-无机杂化钙钛矿电池成为研究开发热点,因其具有稳定的晶体结构及较高的氧化还原性且制造工艺简单、成本低而成为环保型太阳能电池的新选择[4—5].到2020年其太阳能转化率已达到25.2%,研究普遍认为此数据还具有巨大提升空间和广阔前景,从而使钙钛矿电池成为未来新型太阳能电池的主要使用材料[3].
太阳能发电材料是通过吸收光子产生电子-空穴对,使电子跃迁至导带成为自由电子而流动生电,所以入射光能量hν必须大于或等于所用材料的带隙Eg,适当降低带隙值可使光能利用率更高,有利于光能转化.目前,已报道的对CH3NH3PbBr3有关光吸收特性等方面的研究,得出其带隙约为2.274 eV[6].作为光电材料这个带隙值并不在最优区间,限制了光电转化效率;此外,重金属Pb毒性较大,在水体中不可降解,富集过程中对自然环境及生物有巨大危害,这一因素成为大规模应用铅类钙钛矿光电材料的较大限制因素.因此,通过减少Pb的含量降低毒性则是研发安全光电材料的热点之一.而同为过渡金属元素的Bi常温下稳定,毒性远远小于Pb,两者元素表中处于前后位置,其原子相对质量及电荷排布等性质相似,在制备材料中常用Bi对Pb进行替换[7—8].
本文着眼于解决以铅基为主的CH3NH3PbBr3的毒性问题以及将带隙降至最优区间,将B位的部分Pb用Bi代替,对CH3NH3PbBr3进行n型掺杂,通过添加Bi3+离子增加6p电子,使其填充至导带,并实现导带底下移,进而达到减小带隙的目的.运用第一性原理,利用Materials Studio软件中GGA+PBE关联函数,计算不同掺杂量的5种位形得出其能带结构、带隙值、态密度及分波态密度等相关数据,期望得出掺杂状况在形成CH3NH3Pb1-xBixBr3时带隙符合理想结果.
1 CH3NH3Pb1-xBixBr3位形建构
图1a给出了通式为ABX3的正交CH3NH3PbI3钙钛矿晶体结构,空间群:PNMA,空间群号:62,晶格常数a= 8.827 3 Å,b= 12.679 3 Å,c= 8.509 9 Å,α=β=γ=90°[9].有效离子半径较大的I-离子形成八面体网络,八面体顶点即为X位.该八面体存在两种间隙,较大的间隙(A位)被CH3NH3+占据,较小的间隙(B位)被Pb2+占据.通过对I-离子进行Br-离子替代,获得了CH3NH3PbBr3晶体结构(图1b).每个晶胞内含有4个ABX3分子、4个CH3NH3+离子、12个 Pb2+离子、12个 Br-离子.通过对B位的Pb2+进行Bi3+替代,可以获得CH3NH3Pb1-xBixBr3晶体结构位形(图1c).替代比例x可以通过控制替代Pb2+离子的个数实现,文中替代含量分别为x=0, 0.25, 0.50, 0.75, 1.
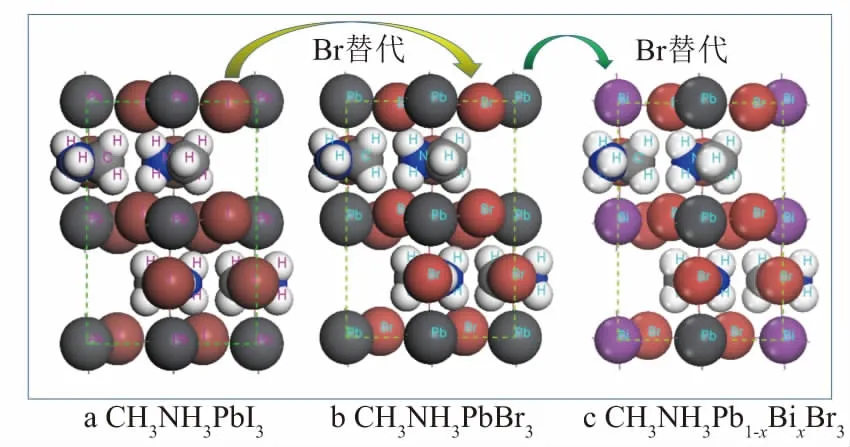
图1 CH3NH3PbI3,CH3NH3PbBr3以及 CH3NH3Pb1-xBixBr3晶体结构位形
2 计算方法
基于密度泛函理论,基态系统的物理性质都由电子密度唯一决定,用电子密度代替波函数,利用Materials Studio中的CASTEP软件包[10—11]计算CH3NH3Pb1-xBixBr3钙钛矿材料的物理性能.计算时选取的C,H,N,Pb,Bi,Br的电子组态分别为2s22p2,1s1,2s22p3,5d106s26p2,5d106s26p3,4s24p5.几何结构优化过程中,对晶格常数a,b,c,α,β,γ均放开弛豫,采用广义梯度近似GGA+PBE[12]作为关联函数及系统推荐的Fine精度.同时考虑自旋极化,采用Koelling-Harmon处理相对论效应,截断能设定为310 eV,能量收敛精度设置为5×10-6eV/atom,最大离子位移设置为0.001 Å,K点选取2×1×2,最大迭代次数设置为1 000次,以保证优化结构到达稳定状态,并获得晶体几何结构参数.采用相同的计算方法对几何优化得到的位形进行计算,获得能带结构、态密度、分波态密度、光学吸收等方面的参数.
3 计算结果与分析
3.1 几何结构分析
表1给出了CH3NH3Pb1-xBixBr3几何优化和能量优化以后得出的晶体结构常数、晶胞体积以及总能.由表1可知,随着Bi含量的增加,晶格常数a与c变化趋势类似,先减小后增加,随后又减小,在x=0.50处获得最大值;而晶格常数b变化趋势与之相反,先增大后减小,再增大,在x=0.25时获得最大值.晶胞体积的变化趋势受到a,b,c共同影响,先增加,而后单调下降,在x=0.25时获得最大值.
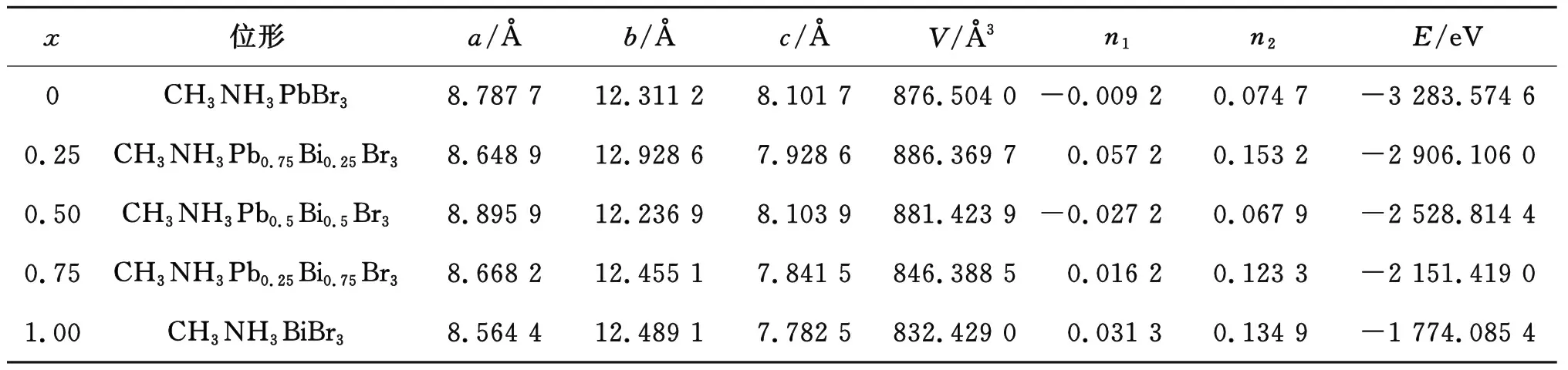
表1 CH3NH3Pb1-xBixBr3 几何结构与系统能量
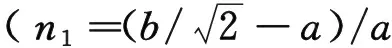
随着Bi含量的增加,系统的总能量呈现线性增加的趋势,其变化规律满足线性变化规律,E(x)=1 509.466 17x-3 283.532 97.内聚能(ECoh)往往用来衡量材料形成的难易程度,可以用下式来表示[15]:
ECoh=ETotal-EC-EN-
6EH-(1-x)EPb-xEBi-3EBr,
(1)
式中:ETotal表示处于基态的晶体能量,eV;EC,EN,EH,EPb,EBi,EBr分别为C,N,H,Pb,Bi,Br孤立原子具有的能量,eV.
图2给出了内聚能随着Bi替代量的变化曲线.由图2可以看到:所有内聚能均为负值,说明所有的结构均有可能形成.然而,随着x的增加,内聚能呈现线性增加的变化规律,且ECoh=3.274 55x-58.695 6.该结果说明Bi3+替代后,材料的稳定性略有降低,试验中对材料进行淬火有可能解决该问题.
3.2 能带结构分析
图3给出了CH3NH3Pb1-xBixBr3能带结构图.
由图3可以看出:
1)当x=0,0.25时,价带顶与导带底同处于Γ点,说明材料为直接半导体材料,带隙分别为2.274,1.564 eV.导带底主要由Pb与Bi的2p电子提供,价带顶主要由Pb,Bi的s电子与Br的p电子杂化共同提供.
2)随着Bi替代量的增加,当x=0.50时,材料由半导体转变为导体,带隙消失,价带穿过费米能级,同时价带与导带存在交叠. Pb与Bi的电子组态分别为5d106s26p2,5d106s26p3,Bi原子比Pb原子多了一个电子,替代过程中多出的电子与Br的p电子杂化,占据更高能级,使带隙降低.当掺杂量达到一定程度时,带隙消失,体系由半导体过渡到导体,该结果与Abdu等使用第一性原理计算的结果类似[16].
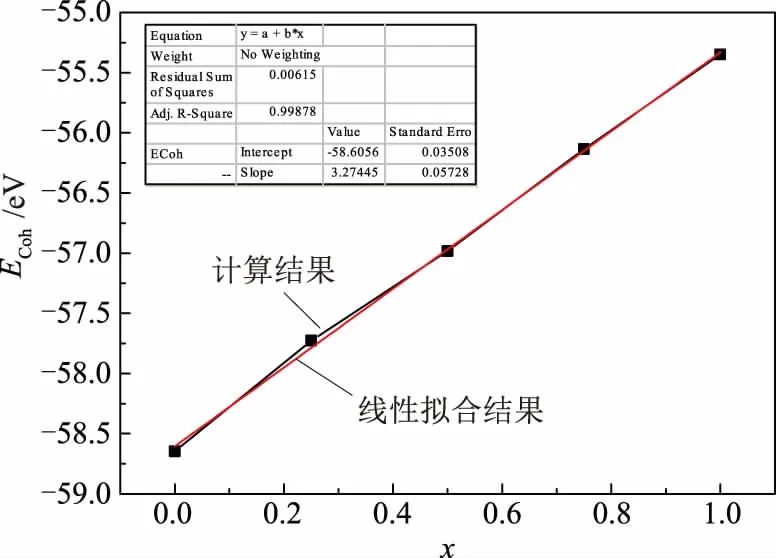
图2 内聚能随Bi含量增加变化关系曲线
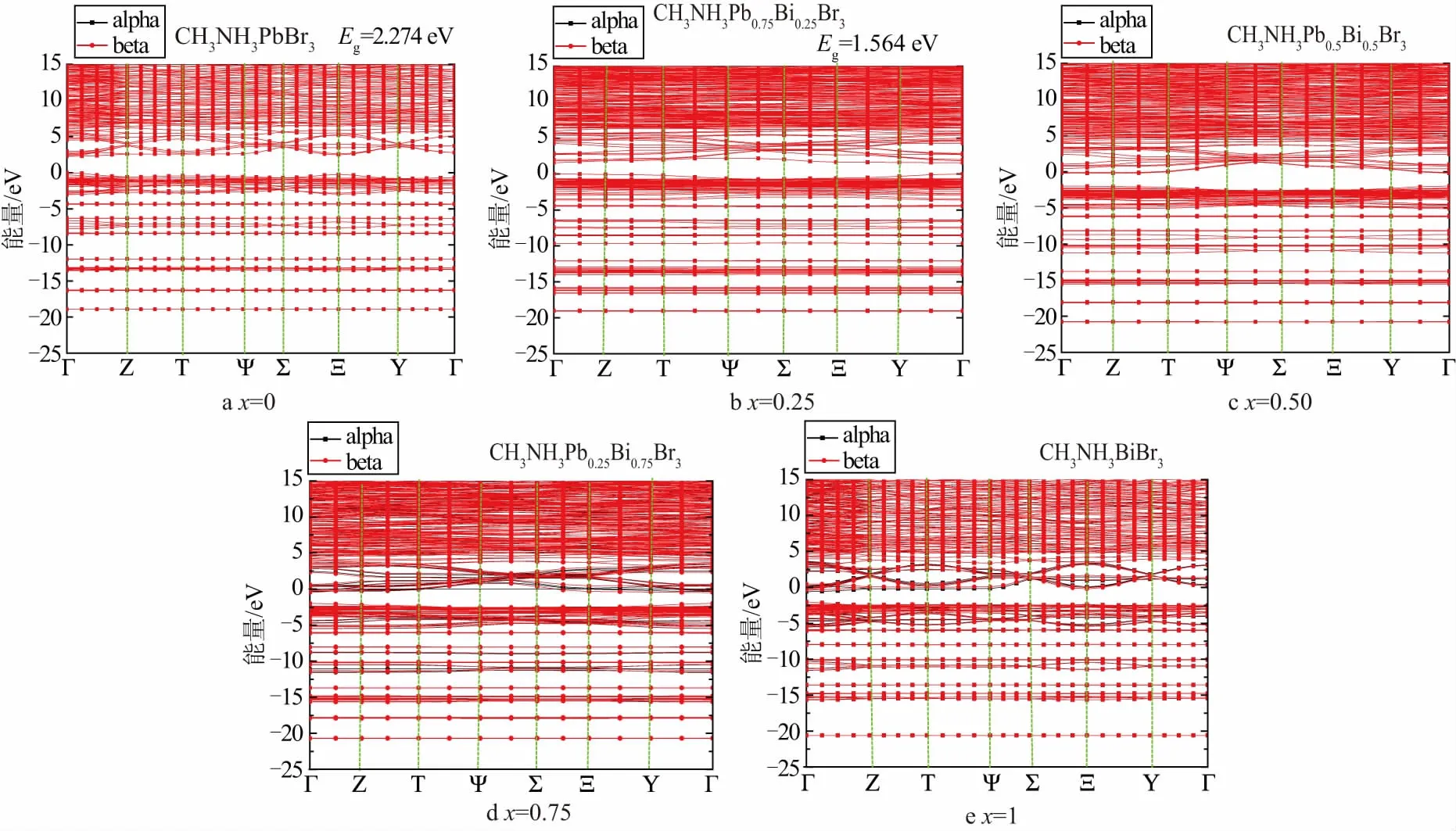
图3 CH3NH3Pb1-xBixBr3能带结构图
3)对于x=0.50,0.75,1.00三个替代含量的结构位形,在费米能级附近的(-1.8 eV,0)能量区间内观察到了空带.随着Bi替代量的增加,载流子浓度不断增大,n型掺杂由初期的浅掺杂逐渐向重掺杂过渡,x=0.50即为浅掺杂极限.
4)电子的有效质量与能带的曲率有关,随着Bi替代量的增加,Γ点附近的能带曲率变化不大,电子的有效质量变化不大,但带隙减小了,这充分说明了Bi的引入加强了电子在价带顶与导带底之间跃迁能力.
图4给出了CH3NH3Pb1-xBixBr3(x=0,0.25,0.50,0.75,1.00)带隙随着Bi替代量变化曲线,由E=hv,c=λv得λ=hc/E,其中:h为普朗克常量,值为4.135 667×10-15eV•s;c为光速;λ≈1 240.7(eV/nm)/Eg(eV)[17].通过改变不同的掺杂量,可以实现材料吸收不同频率的光,即对大于545.602 nm所有的光均可实现吸收.可以看到,Bi替代Pb能够提高材料的光学吸收性能.如果精确控制替代量,可以获得带隙最佳对应的晶体结构.此外,对于x≥0.50的重掺杂结构位形,虽不能作为光电吸收材料使用,但由于其几何结构与低替代量的几何结构类似,可以作为缓冲层来使用,以此降低界面缺陷,有助于提高光电转化效率.

图 4 CH3NH3Pb1-xBixBr3带隙随Bi替代量变化曲线
3.3 态密度分析
图5给出了CH3NH3Pb1-xBixBr3的5种位形态密度图,由图5可以看出:
1)CH3NH3PbBr3,CH3NH3Pb0.75Bi0.25Br3的位形在费米能级处没有电子态,故这两种材料为半导体,费米能级左侧为价带,右侧为导带,与能带结果获得结果一致.而对于CH3NH3Pb0.5Bi0.5Br3,CH3NH3Pb0.25Bi0.75Br3,CH3NH3BiBr3三种位形,导带与价带交叠在一起,呈现明显的半金属特性.

图 5 CH3NH3Pb1-xBixBr3 整体态密度图
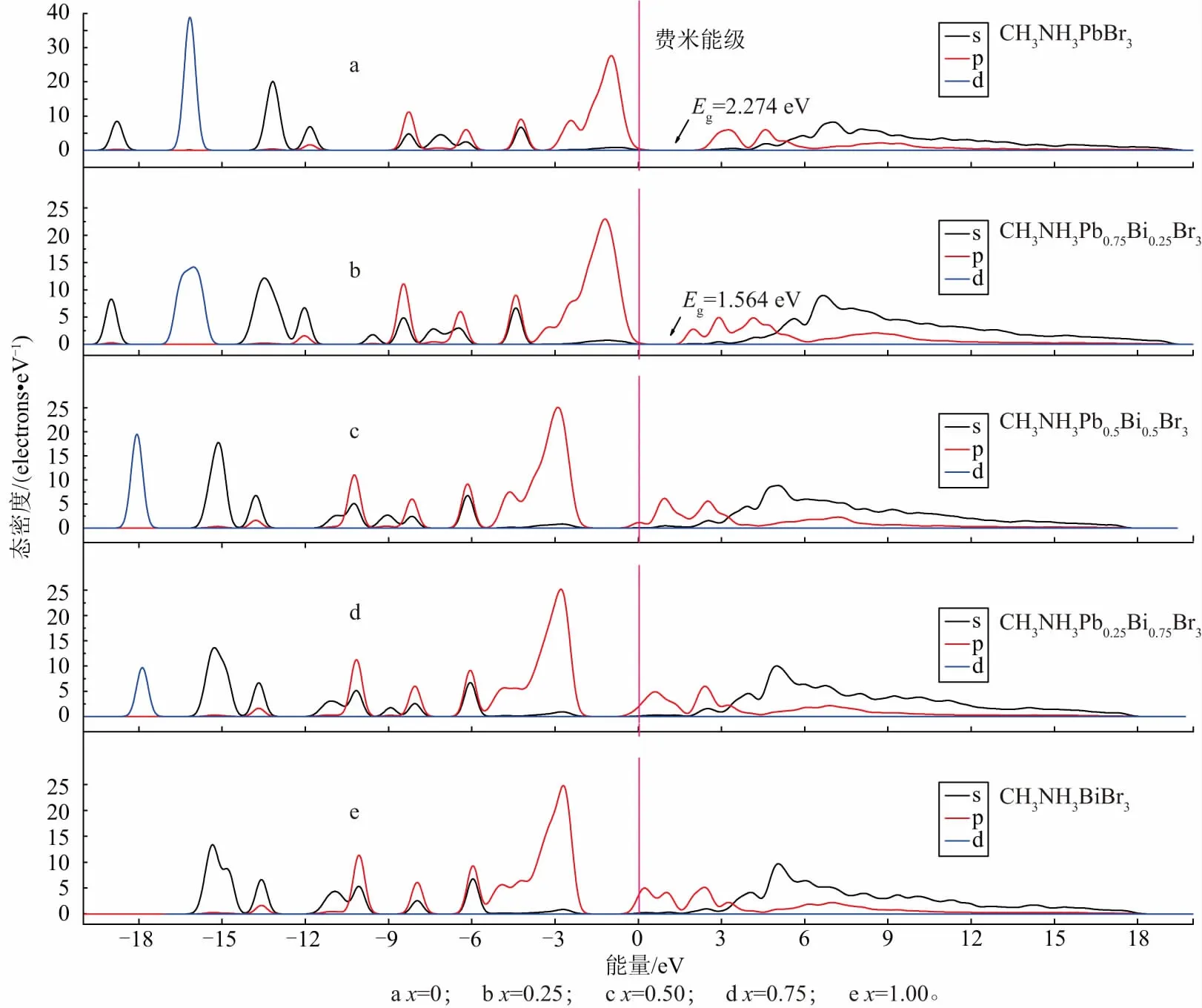
图 6 CH3NH3Pb1-xBixBr3 分波态密度图
2)在费米能级以下,随着Bi替代量的增加,电子状态分布的能量区间逐渐收窄,其主要原因是导带整体向低能量移动,并与价带在费米能级附近交叠.
3)如果定义费米能级附近的两个尖峰峰值间的能量插值为“赝能隙”[18],随着Bi含量的增加,“赝能隙”逐渐变窄.这说明共价键成键能力逐渐减弱,离子键成键能力不断增强.
3.4 分波态密度分析
图6分别给出了 CH3NH3Pb1-xBixBr3分波态密度图.对于s电子:在费米能级附近,Pb,Bi的s电子与Br的p电子杂化明显,决定了价带的主要性质.随着Bi替代量的增加,其变化趋势与总态密度变化类似,而s电子的变化主要来自于Bi的替代,n型掺杂增加了s电子浓度,说明了Bi替代Pb的主要作用之一来自于s电子对整体态密度分布的调控,提高了价带顶的位置.对于p电子:在(-11.3,8.7)能量区间范围内,p层电子分布最多,Bi与Pb的6p电子占据导带底,与s,d电子基本没有杂化,在费米能级处相互作用明显,决定了导带的主要性质.增加Bi含量实质是增加6p电子,同时占据新的能级,降低导带底的位置.当Bi替代含量为0.50时,占据的新能级穿过费米能级进行使材料形成导体.对于d电子而言,电子状态位于远离费米能级的价带位置,态密度峰较尖,电子相对局域化,对应能带较窄,有效电子质量较大,对材料性能影响不大.
3.5 Bi替代物理机理分析
通过3.3节与3.4节的分析与讨论可知, CH3NH3Pb1-xBixBr3位形的价带顶主要由Pb,Bi的s电子与Br的p电子杂化后共同占据,导带底则由金属离子的p电子占据.由于Bi(6s2p3)比Pb(6s2p2)多了一个p电子,排除几何结构的影响,可以将其主要作用简化为在体系中增加6p电子.图7给出了Bi取代Pb调控带隙物理机制示意图.由图7可见,随着Bi替代量的增加,主要决定导带底的6p电子逐渐增多,6p将会填充到靠近导带底位置的新能级(如黄色能级所示),进而导致带隙降低;随着替代量的进一步增加,6p电子数量增多,母体带隙部分的能级不足以填充这些6p电子,6p电子只能越过费米能级,进一步向低能级占据,导致价带与导带交叠,形成半金属特性.
4 结论
根据第一性原理,利用Materials Studio中的CASTEP软件包构建出CH3NH3Pb1-xBixBr3(x=0,0.25,0.50,0.75,1.00)5种位形,使用GGA+PBE作为关联函数进行位形的几何优化,得出稳定结构后再计算分析其能带结构、态密度、分波态密度等物理性质,得出以下结论:
1)随着对母体替代Bi3+离子含量的增加,晶胞体积先增加后单调减小,由内聚能分析发现所有位形均可形成,但难度逐渐增加.
2)Bi替代实现了CH3NH3Pb1-xBixBr3半导体-金属转变.通过精细调节替代含量,可以获得符合可见光吸收的CH3NH3Pb1-xBixBr3有机无机杂化钙钛矿材料,其特点是降低了Pb的使用含量.
3)Bi替代的主要作用是通过p电子调控导带底能级实现的,进而提出了Bi取代Pb调控带隙的物理机制.

图 7 Bi取代Pb调控带隙物理机制示意图
