香灰菌gpd启动子的克隆及功能验证
2019-09-23刘冬梅许丹云王媛媛马爱民
陈 悦,刘冬梅,许丹云,王媛媛,马爱民
(华中农业大学食品科学技术学院,湖北武汉 430070)
香灰菌(Hypoxylonsp.)是一种非致病性真菌,作为银耳的伴生菌,它能够分解基质,为银耳提供丰富的营养物质,促进银耳的生长发育[1]。香灰菌在生长过程中会产生并分泌大量的天然黑色素,具有抗氧化能力,能够提高多酚氧化酶的活力[2],可应用于食品及饮料色泽、品质的改善[3]。香灰菌产生的挥发性有机化合物(VOCs),包括烷烃类、烯烃类、酮类、芳香族化合物等,可作为生物燃料、天然风味化合物、抗真菌剂等[4-5],有研究表明,香灰菌产生的挥发性有机化合物中含有桉油精,能显著抑制番茄上尖孢镰刀菌的生长[6],在果蔬等农产品的保藏中具有一定的应用价值。
为了构建香灰菌的转化体系,研究香灰菌的基因功能,需要获得高活性的启动子元件。3-磷酸甘油醛脱氢酶(glyceraldehyde-3-phosphatedehydrogenase,GPD)是糖酵解和糖异生途径中的关键酶,其mRNA占酵母中poly(A)+RNA的2%~5%,表明gpd基因受高活性启动子调控[7-8]。gpd启动子是一个组成型表达的强启动子,不依赖外界环境诱导,可持续驱动下游基因的表达[9],研究发现,内源启动子能显著提高真菌的转化效率[10-11],所以许多研究都从各自的材料中分离启动子。目前,gpd启动子已相继从不同的担子菌如糙皮侧耳(Pleurotusostreatus)[11]、裂褶菌(Schizophyllumcommune)[12]、草菇(Volvariellavolvacea)[13]、金针菇(Flammulinavelutipes)[14]和子囊菌如构巢曲霉(Aspergillusnidulans)[15]、沙门柏干酪青霉(Penicilliumcamemberti)[16]、玉米丝轴黑粉菌(Sporisoriumreilianumf. sp.zeae)[17]、立枯丝核菌(Rhizoctoniasolani)[18]、烟曲霉(Aspergillusfumigatus)[19]等真菌中成功分离,但是目前还没有从香灰菌中克隆到gpd启动子的报道。
本实验通过聚合酶链式反应(polymerase chain reaction,PCR)获得香灰菌gpd上游序列,利用生物信息学分析及实验验证其启动子的功能,这将为后续香灰菌基因功能的研究和外源蛋白的表达奠定基础。
1 材料与方法
1.1 材料与仪器
香灰菌(Hypoxylonsp.) 分离自福建三明真菌所提供的银耳香灰混合菌种;pCAMBIA1302-pgpd-egfp-hph质粒 改造自载体pCAMBIA1302,载体T-DNA所含的hph基因和egfp基因由糙皮侧耳gpd启动子驱动,华中农业大学食品微生物实验室构建[20];大肠杆菌DH5α、根癌农杆菌GV3101感受态、TaqDNA polymerase、dNTPs、Trans2K plus II DNA Marker 北京全式金生物技术有限公司;限制性内切酶、pMD18-T克隆载体及Solution I、DNA Ligation Mix、cDNA合成试剂盒Prime ScriptTMRT reagent Kit with gDNA Eraser 大连TaKaRa;凝胶纯化试剂盒、胶回收试剂盒和质粒提取试剂盒 美国Axygen;PDA及PDB培养基 美国Difco;YEB培养基、50×TAE电泳缓冲液 国药集团化学试剂有限公司;氨苄青霉素(Amp)、卡那霉素(Kan)、头孢噻肟霉素(Cefo)、利福平(Rif)、乙酰丁香酮(AS) 美国Sigma-Aldrich;潮霉素B(Hyg B) 上海翊圣生物科技有限公司;诱导培养基(IM)及共培养培养基(Co-CM)配制方法:0.8 mL 1.25 mol/L K-Buffer(pH 4.8)、20 mL MN-Buffer、1 mL 1% CaCl2·2H2O(w/v)、10 mL 0.01% FeSO4(w/v)、2.5 mL 20% NH4NO3(w/v)、10 mL 50%甘油(v/v)、5 mL trace elements(100 mg/L ZnSO4·7H2O、100 mg/L CuSO4·5H2O、100 mg/L H3BO3、100 mg/L Na2MoO4·2H2O)、40 mL 1 mol/L 2-N吗啉代乙磺酸(MES)(pH 5.5)、20% 葡萄糖(w/v)(IM中10 mL,Co-CM中则为5 mL),用H2O补齐至1000 mL;MES 美国Amresco;其余试剂 国药集团化学试剂有限公司。
T100TMThermal Cycler PCR仪、Gene Pluser XcellTM电穿孔系统 美国Bio-Rad公司;DU2640型核酸分析仪 美国Beckman Coulter;Eclipse80i正置荧光显微镜 日本Nikon;Gel Logical 200凝胶成像系统 美国Kodak;Centrifuge 5415R冷冻高速离心机 德国Eppendorf;DYY-8C型电泳仪 北京六一仪器厂。
1.2 实验方法
1.2.1 基因组DNA和总RNA的提取 用无菌接种针从PDA斜面培养基中挑取香灰菌丝,接种于PDA培养基中,25 ℃培养5 d,活化菌种。将活化好的菌丝接种于贴有玻璃纸的PDA培养基中,25 ℃培养7 d,用于DNA及RNA的提取。香灰菌基因组DNA提取采用改进的CTAB法[20],总RNA的提取采用改进的Trizol法[21],用0.8%琼脂糖凝胶电泳分别检测DNA及RNA样品的完整性,用DU2640型核酸分析仪分别检测DNA及RNA样品的浓度及纯度,并保存于-80 ℃冰箱中,用于后续操作。
1.2.2 菌株gpd启动子重组质粒的构建
1.2.2.1gpd基因片段及其启动子的克隆 根据JGI数据库(https://genome.jgi.doe.gov/portal/)中的香灰菌(Hypoxylonsp. CO27)3-磷酸甘油醛脱氢酶基因序列[4],分析gpd基因序列的同源性,根据gpd基因序列保守区域,利用Primer 5.0软件分别设计引物序列H1-gpd-F(5′-CGTCTTCCGCAATGCTGTT-3′)和H1-gpd-R(5′-CGAAGTTCTTGTTCAGGGAGA-3′)。根据试剂盒要求将RNA稀释至适宜浓度后,第一链cDNA的合成采用Prime ScriptTMRT reagent Kit with gDNA Eraser(TAKARA)试剂盒,操作参照试剂盒说明书。以香灰菌cDNA为模板扩增gpd基因片段,PCR体系为10×Buffer 2.0 μL、dNTPs 0.8 μL、引物F 1 μL、引物R 1 μL、模板1 μL、TaqDNA polymerase 0.2 μL、ddH2O 14 μL(以下PCR体系均参照本体系)。PCR扩增条件为:95 ℃预变性3 min,95 ℃变性30 s,60 ℃复性30 s,72 ℃延伸1 min,30个循环后72 ℃延伸10 min,4 ℃保存。用1.0%琼脂糖凝胶电泳检测并分离目的条带,切胶回收产物与pMD18-T载体于16 ℃连接2 h后,转化大肠杆菌DH5α感受态细胞中,100 μg/mL氨苄青霉素抗性筛选及菌落PCR验证,选择阳性克隆子送天一辉远公司(武汉)进行测序。
将已经克隆得到的序列与JGI数据库进行比对分析,选择同源性最高的序列为模板,设计引物promoter-F(5′-GAACCCGTTTGACATTCGTG-3′)与引物H1-gpd-R扩增gpd基因片段及其上游部分,以香灰菌DNA为模版进行PCR扩增,PCR体系同上,PCR扩增条件为:95 ℃预变性3 min,95 ℃变性30 s,58 ℃复性30 s,72 ℃延伸2 min,30个循环后72 ℃延伸10 min,4 ℃保存,后续步骤同上,得到gpd启动子与基因连续的片段。
1.2.2.2gpd启动子区域的分析gpd启动子借助Neural Network Promoter Prediction(http://www. fruitfly.org/seq_tools/promoter.html)软件、Signal Scan Search Request(https://www.arduino.cc/en/Tutorial/GSMToolsGsm ScanNetworks)、PLANTCAREA database(http://bioinformatics.psb.ugent.be/webtools/plantcare/ html/)AliBaba2.1(http://gene-regulation.com/pub/programs/alibaba2/index.html?)及NCBI Blast(https: //blast.ncbi.nlm.nih.gov/Blast.cgi)进行预测分析。
1.2.3 香灰菌gpd启动子功能验证
1.2.3.1 香灰菌gpd启动子表达载体构建 通过DNAMAN V6软件分析克隆得到的香灰菌gpd启动子的序列及初始载体pCAMBIA1302-pgpd-egfp-hph序列发现,香灰菌gpd启动子不含有EcoR I和BamH I位点,但是初始载体含有两个BamH I酶切位点,不可以使用EcoR I和BamH I双酶切直接将pgpd启动子替换,所以选择位于hph片段下游的酶切位点XhoI,分步克隆带有酶切位点的香灰菌hgpd启动子及egfp-hph片段。将设计带有酶切位点EcoR I和BamH I的特异引物H-EcoR I-F(5′-GGAATTCGATGTCTGCTCGCCTCG-3′)H-BamH I-R(5′-GGGATCCGATGCCAACCTTGACGA-3′)(横线处为酶切位点),用PCR法扩增得到带有EcoR I和BamH I酶切位点的香灰菌gpd启动子片段。用引物BamH I-egfp-F(5′-CGGATCCTGAGCAAGGGCGAG-3′)和XhoI-hph-R(5′-CCTCGAGCGGCTATTCCTTT GCCCTCG-3′)(横线处为酶切位点)扩增带有BamH I和XhoI酶切位点的egfp和hph片段,PCR体系为10×Buffer 2.0 μL、dNTPs 0.8 μL、引物F 1 μL、引物R 1 μL、模板1 μL、TaqDNA polymerase 0.2 μL、ddH2O 14 μL。PCR扩增条件为:95 ℃预变性3 min,95 ℃变性30 s,65 ℃复性30 s,72 ℃延伸1 min,30个循环后72 ℃延伸10 min,4 ℃保存。分别酶切gpd片段、egfp-hph片段,抽提本实验室已有的pCAMBIA1302-pgpd-egfp-hph质粒,用EcoR I和XhoI双酶切,与带有酶切位点的gpd启动子和egfp-hph片段,琼脂糖凝胶电泳检测各片段的相对浓度,按一定的比例用DNA Ligation Mix在16 ℃条件下连接12 h,构建表达载体pCAMBIA1302-hgpd-egfp-hph,载体结构如图1所示。
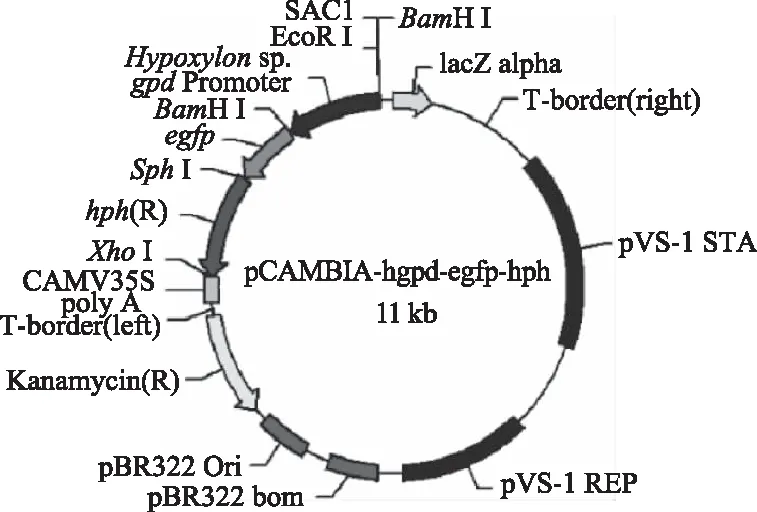
图1 pCAMBIA1302-hgpd-egfp-hph载体图Fig.1 pCAMBIA1302-hgpd-egfp-hph vector map
1.2.3.2 根癌农杆菌的电击转化 提取表达载体pCAMBIA1302-hgpd-egfp-hph质粒,在无菌条件下将0.1~0.5 μg纯化的质粒加入根癌农杆菌感受态细胞中,将混合液转移至预冷的电击杯(电极间距1 mm)中,并置于电脉冲仪电极之间,在2.5 kV 25 mF 200 ohms条件下电击5 ms,取出电击杯,加入1 mL无抗的YEB培养基,转移至1.5 mL离心管中,28 ℃ 200 r/min振荡培养约3~4 h,然后涂布于50 μg/mL利福平和50 μg/mL卡那霉素的YEB抗性平板,28 ℃培养2 d,利用菌落PCR方法筛选根癌农杆菌,使用H-EcoR I-F、XhoI-hph-R引物,以根癌农杆菌单菌落为模板,以抽提出的pCAMBIA1302-hgpd-egfp-hph质粒为阳性对照,PCR体系为10×Buffer 2.0 μL、dNTPs 0.8 μL、引物F 1 μL、引物R 1 μL、模板1 μL、TaqDNA polymerase 0.2 μL、ddH2O 14 μL,PCR扩增条件为95 ℃预变性3 min,95 ℃变性30 s,58 ℃复性30 s,72 ℃延伸3 min,30个循环后72 ℃延伸10 min,4 ℃保存。
1.2.3.3 根癌农杆菌介导香灰菌丝的转化 生长于PDA平板上的香灰菌丝体在25 ℃条件下培养5 d后,用直径6 mm打孔器均匀打取平板边缘的菌丝块,分别接种于含有潮霉素浓度50、100、110、120、130、140、150 μg/mL和含有头孢噻肟霉素浓度为200 μg/mL的PDA选择平板的中央,25 ℃避光培养,观察菌丝的生长状况。
将香灰菌丝在PDB液体培养基中活化5 d,含有重组质粒的根癌农杆菌在含有50 μg/mL利福平和50 μg/mL卡那霉素的YEB液体培养基中活化,用含有200 μg/mL乙酰丁香酮的诱导培养IM[22]培养根癌农杆菌,诱导根癌农杆菌毒性,当OD600达到0.4~0.6时,用根癌农杆菌与磨碎的菌丝碎片等体积混合,然后将混合物涂布在附有微孔滤膜(0.45 μm,60 mm)的Co-CM固体培养基上(含200 μg/mL乙酰丁香酮)[23]。然后将根癌农杆菌与菌丝体共培养,25 ℃条件下培养3 d后,将菌丝体转移至含有150 μg/mL潮霉素和200 μg/mL头孢噻肟霉素的选择性平板上25 ℃培养7~10 d,待转化子萌发。
1.2.3.4 拟转化子的筛选和验证 将选择性平板上长出的菌丝体转接至新的选择培养基上,在潮霉素抗性选择的压力下部分菌丝生长较快,将生长较快的菌丝接种到新的选择性平板上,仍然快速生长的菌丝接种至贴有玻璃纸的无抗PDA平板上生长,再次转接至选择平板仍能生长的菌丝即为具有稳定潮霉素抗性的拟转化子。将筛选出的拟转化子接种到贴有玻璃纸的无抗PDA平板上,用CTAB法提取拟转化子的基因组DNA,对拟转化子进行PCR验证,以DNA为模板用hph-F(5′-CTCGTGCTTT CAGCTTCGATGTAGG-3′)和hph-R(5′-CGGTTT CCACTAT CGGCGAGTACTT-3′)为引物进行PCR扩增,PCR体系为10×Buffer 2.0 μL、dNTPs 0.8 μL、引物F 1 μL、引物R 1 μL、模板1 μL、TaqDNA polymerase 0.2 μL、ddH2O 14 μL,反应条件为:95 ℃预变性3 min,95 ℃变性30 s,65 ℃复性30 s;72 ℃延伸1 min,30个循环后72 ℃延伸10 min,4 ℃保存。
1.2.3.5 荧光蛋白在香灰菌转化子中的表达 随机选取两株转化子,挑取这些转基因菌株的菌丝接种于含有潮霉素(150 μg/mL)的抗性平板上,在菌落旁插入无菌盖玻片使菌丝爬片生长,待菌丝覆盖盖玻片一半面积时,小心取出盖玻片,菌丝覆盖面朝下将盖玻片压在载玻片上。做好标记后,使用荧光显微镜,选择B-2A型滤镜通过蓝光激发,分别在10×、20×物镜下观察菌丝细胞中绿色荧光蛋白表达情况。用随机软件NIS-Elements BR3.0(Nikon,Japan)进行标注分析。
2 结果与分析
2.1 香灰菌gpd启动子的克隆
香灰菌丝总DNA和RNA的电泳胶图如图2所示,条带表明DNA和RNA都比较完整,无明显降解,符合PCR扩增要求。gpd基因部分片段cDNA和DNA序列扩增结果如图3A所示,
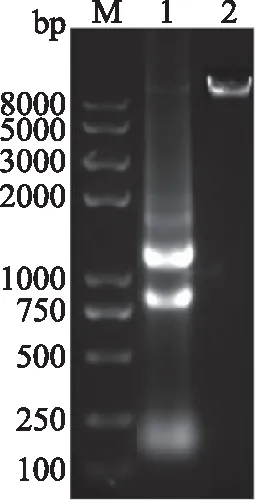
图2 香灰菌总RNA与DNAFig.2 Total RNA and DNA of Hypoxylon sp.注:M:Trans 2K DNA Marker; 泳道1为RNA,泳道2为DNA。
条带大小符合预期且无杂带。测序结果显示,cDNA模板扩增出873 bp片段,DNA模板扩增出933 bp片段,基因片段中有一个60 bp的内含子区域。然后将序列与NCBI(https://www.ncbi.nlm.nih.gov/)Blastx比对,经分析该序列与Nodulisporiumsp. 65-12-7-1(KY977747.1)同源性达94%,内含子所在位置也相近,表明已经获gpd基因片段。根据同源性最高的序列为模板,设计引物,扩增gpd启动子与基因连续的片段,得到一条1589 bp的序列,如图3B所示。

图3 gpd基因部分片段及其启动子片段扩增产物Fig.3 Partial fragment of gpd gene and its promoter fragment amplification product
2.2 香灰菌gpd启动子序列分析
gpd基因的上游侧翼序列经Neural Network Promoter Prediction软件和Signal Scan Search Request软件进行预测分析,结果显示,gpd启动子功能序列分布于克隆的gpd片段的上游1000 bp区域内,启动子包含一个高分值起始转录位点,Neural Network Promoter Prediction软件预测在ATG上游23 bp处有一个分值高达0.99分的预测转录起始位点,PLANTCAREA database软件分析该启动子包含完整的CAAT-box、GATA-box、TATA-box、GC-box等核心元件及一些顺式作用元件如A-box、NIT2 motif等。在启动子核心元件的远上游区域有两个典型的CAAT-box,如图4所示,该结构与糙皮侧耳[24]和草菇[25]gpd启动子CAAT-box所处位置类似。AliBaba 2.1软件分析出该启动子序列含有许多转录因子结合位点,例如,HSF motif热调控元件,GATA-box增强子,TCT-motif、L-box和SP1光响应元件等顺式作用元件。
2.3 香灰菌gpd启动子功能的验证
2.3.1 表达载体的构建 琼脂糖凝胶检测各片段的相对亮度结果如图5A所示,pCAMBIA1302-pgpd-egfp-hph载体骨架浓度比gpd启动子片段和egfp-hph片段低,所以将连接体系设置为5 μL DNA Ligation Mix、2 μL pCAMBIA1302-pgpd-egfp-hph载体骨架、1.5 μLgpd启动子片段和1.5 μLegfp-hph片段,连接后,转化大肠杆菌DH5α,菌落PCR用H-EcoR I-F和XhoI-hph-R验证,扩增出2700 bp条带,与预期大小相符,表明融合表达载体pCAMBIA1302-hgpd-egfp-hph构建成功。如图5B所示,电击转化根癌农杆菌GV3101,涂布于50 μg/mL利福平和50 μg/mL卡那霉素的YEB抗性平板,菌落PCR结果与质粒阳性对照一致,表明转化根癌农杆菌成功。
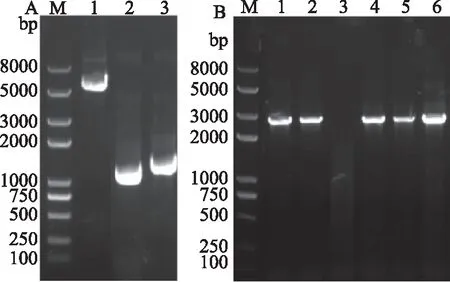
图5 pCAMBIA1302-hgpd-egfp-hph载体构建过程Fig.5 pCAMBIA1302-hgpd-egfp-hph plasmid construction process
2.3.2 拟转化子的筛选及验证 将野生型菌株在25 ℃培养箱中避光培养,观察菌丝的生长状况,在140 μg/mL的潮霉素选择性平板上仍能缓慢生长,150 μg/mL潮霉素选择性平板上不生长,所以选择150 μg/mL作为转化子筛选的抗性浓度。而在200 μg/mL头孢噻肟霉素选择性平板上生长状况与无抗生素平板一致,表明头孢噻肟霉素对香灰菌生长不影响,可以用于转化子筛选时抑制根癌农杆菌的生长。
香灰菌丝经根癌农杆菌侵染后,在潮霉素抗性选择的压力下部分菌丝生长较快,其余呈现死亡状态,筛选三代后仍然有生长较好的菌丝,初步说明菌丝被转化成功。随机挑选拟转化子,提取DNA作为模板扩增hph基因,并用野生型作为阴性对照,质粒作为阳性对照,结果如图6所示,在840 bp位置得到特异性条带,表明菌丝体内存在外源hph基因。
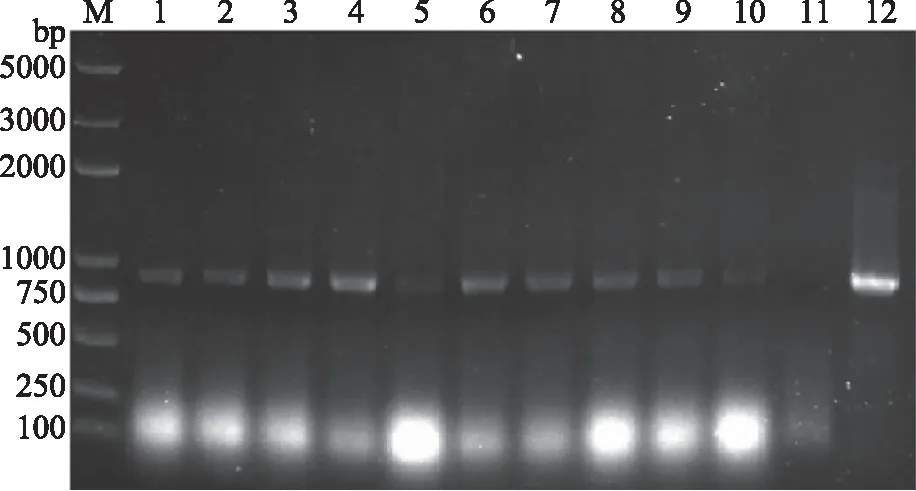
图6 转化子PCR的检测结果Fig.6 PCR detection results of transformants
随机挑取两株转化子爬片生长好的菌丝制好片后,用荧光显微镜观察,从图7中可以看出荧光显微镜中能观察到明显的绿色荧光,香灰菌gpd启动子能有效驱动egfp的表达,同时也表明根癌农杆菌介导的香灰菌丝转化成功。
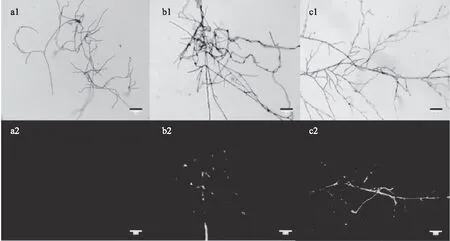
图7 不同转化子菌株中绿色荧光蛋白表达检测结果Fig.7 Detection results of green fluorescent protein expression in different transformant strains注:a1:明场中的野生型菌株;a2:荧光下的野生型菌株;b1、c1为随机挑选的 两株转化子菌株在明场中的照片,b2、c2为转化子菌株在荧光下的照片。
3 结论
本试验利用PCR技术克隆得到香灰菌gpd基因上游1000 bp的DNA片段,通过生物信息学分析,该片段具有典型的启动子特征,序列中除了包含一个高分值的转录起始位点,也含有增强子CAAT-box、核心启动元件TATA-box,在核心元件TATA-box与转录起始位点之间有一个对驱动效率有重要影响的富含CT的区域。此外,还含有G-box、GC-box、HSF motif、L-box、SP1等顺式作用元件。进一步将该片段与潮霉素抗性基因和绿色荧光蛋白基因连接构建表达载体,再利用根癌农杆菌介导转化香灰菌,结果显示,该片段能够驱动潮霉素抗性基因和绿色荧光蛋白基因在香灰菌中稳定表达,说明所得片段的确为香灰菌的gpd启动子,该启动子能够用于今后香灰菌基因表达及基因功能的研究。
