呋喃鬼臼毒素衍生物及其抗肿瘤活性研究
2016-11-22成伟华牛聪孙强稳陈虹邹忠梅
成伟华,牛聪,孙强稳,陈虹,邹忠梅
(1.中国医学科学院北京协和医学院药用植物研究所,北京100193;2.原子高科股份有限公司,北京102413;3.武警总医院医务部,北京100039;4.武警8720部队医院药械科,无锡214000;5.武警后勤学院生药学教研室,天津300309)
论著
呋喃鬼臼毒素衍生物及其抗肿瘤活性研究
成伟华1,2,牛聪3,孙强稳4,陈虹5,邹忠梅2
(1.中国医学科学院北京协和医学院药用植物研究所,北京100193;2.原子高科股份有限公司,北京102413;3.武警总医院医务部,北京100039;4.武警8720部队医院药械科,无锡214000;5.武警后勤学院生药学教研室,天津300309)
目的:获得抗肿瘤活性更强的鬼臼毒素衍生物。方法:以鬼臼毒素为起始原料,通过把取代呋喃和鬼臼毒素拼接,设计并合成5个4β-N-取代呋喃鬼臼毒素衍生物,目标产物的结构通过1H-NMR,13C-NMR和HRMS的确证,同时采用MTT法评价新化合物对HeLa,K562和K562/A02的细胞毒活性。结果:合成的5个化合物均具有确切的细胞毒活性,其中化合物7c和11b对于耐药肿瘤细胞K562/A02的活性明显优于阳性对照依托伯苷。结论:把取代呋喃连接到鬼臼毒素可以增加抗肿瘤活性。
鬼臼毒素;结构修饰;抗肿瘤活性.
癌症严重威胁人类的健康和生命,已成为人类第一位的致死性病因,现有的肿瘤化学治疗药物存在疗效低、毒性大、难于吸收、价格昂贵及易于产生耐药性等缺点,因此,全球每年投入大量的人力及物力寻找和开发防治癌症的新药[1]。鬼臼毒素是从鬼臼属(Dysosma)植物中分离得到的具有显著抗肿瘤活性的芳基四氢萘类木脂素内酯。由于鬼臼毒素的毒性较大在应用上受到很大的限制,此后经过结构修饰合成得到鬼臼毒素衍生物依托泊苷(VP-16)和替尼泊苷(VM-26),并成为抗肿瘤临床药物[2]。但它们在实际临床使用中却常常受到限制,其中最主要的问题是水溶性差和存在骨髓抑制等毒副作用[3-4]。构效关系研究表明保留反式内酯环的基础上,C-4位是比较有效的修饰位点,C-4位可以通过氧、硫、氮原子来连接。一般而言,以氮原子连接的衍生物的活性都比较好,而4β-氮取代鬼臼毒素衍生物对多种人类癌细胞株显示出比VP-16还要高的抗癌活性[5-7]。为进一步寻找更为高效和低毒的抗肿瘤新药,本研究分析鬼臼毒素类化合物构效关系,在保留C-4位取代基的β构型及内酯环的反式取向的基础上,集中在C-4位上设计合成了一系列4β-氮取代鬼臼毒素衍生物,其结构经1H-NMR,13C-NMR和HRMS的确证,并经MTT法筛选了该类衍生物的体外抗肿瘤和抗多药耐药活性。
1 仪器与方法
1.1仪器与试剂熔点测定仪:Fisher-Johns熔点仪(温度计未校正)。核磁共振光谱用BRUKERBRUKER ARX-600型核磁仪测定,采用CDCl3为溶剂,四甲基硅烷(TMS)为内标,偶合常数(J)以赫兹(Hz)为记录单位。高分辨质(HR-ESI-MS)在Quattro MicroTM API(Waters,Milford,MA)质谱仪上测定,比旋光度在Perkin-Elmer 241 MC旋光仪上于室温(25℃)测定,光源选择钠灯D线。薄层色谱(TLC)(GF254,0.5 mm,青岛产),柱层色谱硅胶:60-100目,100-200目,200-300目均为青岛海洋化工厂产品,薄层层析硅胶:GF 254(青岛海洋化工厂)。MQX-200酶标仪:Bio-Tek.Instruments Inc.,USA。鬼臼毒素(98%)购自南京青泽医药科技开发有限公司。若无特殊说明,实验试剂均为市售化学纯或分析纯,未经说明没有另外纯化。
1.2合成路线本路线采用糠醇为起始原料,首先在冰醋酸条件下与R2NR3发生Mannich反应生成N-取代呋喃中间体,然后经过氧化生成N-取代呋喃甲醛中间体3a-3c(图1),该中间体在常温条件下与4β-氨基-4-脱氧表鬼臼毒素或4β-氨基-4′-去甲基-4-脱氧表鬼臼毒素发生反应后,再经还原反应生成目标化合物7a-7c和11a-11b(图2)。
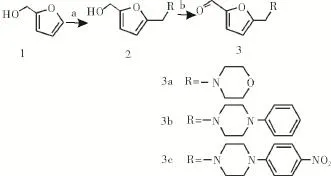
图1 化合物3a-3c的合成路线Fig 1 Synthesis of com pound 3a-3c
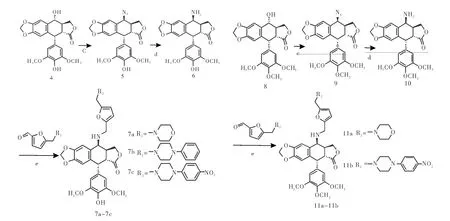
图2 化合物7a-7c, 11a-11b的合成路线Fig 2 Synthesis of com pound 7a-7c, 11a-11b
合成中间体4β-氨基-4-脱氧表鬼臼毒素采用在三氟乙酸条件下,鬼臼毒素与叠氮化钠发生取代反应制得中间体4β-叠氮基-4-脱氧表鬼臼毒素,中间体4β-叠氮基-4-脱氧表鬼臼毒素在10%Pd/C催化下被甲酸铵还原为4β-氨基-4-脱氧表鬼臼毒素[8-9]。
1.3合成实验
1.3.1鬼臼毒素类衍生物中间体的合成
1.3.1.1 4β-氨基-4-脱氧表鬼臼毒素10的合成:将鬼臼毒素8(4.14 g,10 mmol)溶于50 mL干燥二氯甲烷中,小心加入NaN3(2.60 g,40 mmol)搅拌使其溶解,0℃下将10 mL CF3COOH缓慢滴加到反应液中,反应1 h后,室温反应4 h。TLC检测反应完成,0℃下,滴入NaHCO3饱和溶液至无气泡为止,分出有机层,无水Na2SO4干燥,减压蒸干,粗品用二氯甲烷:乙酸乙酯(1∶1)重结晶,得白色晶体4β-叠氮-4-脱氧表鬼臼毒素9 3.99 g,收率89%。将4β-叠氮-4-脱氧表鬼臼毒素9(4.39 g,10 mmol)溶于50 mL乙酸乙酯中,加入10%Pd/C(1.00 g),HCOONH4(2.52 g,40 mmol),加热回流搅拌5 h,TLC检测反应完全,过滤回收Pd/C,滤液用饱和食盐水洗3遍,减压蒸干得白色泡沫状固体粗产物4β-氨基-4-脱氧表鬼臼毒素10 3.63 g,收率88%,不经进一步分离纯化可直接用于下一步反应[10]。
1.3.1.2 4β-氨基-4′-去甲基-4-脱氧表鬼臼毒素6的合成:将4′-去甲基鬼臼毒素4(4.00 g,10 mmol)溶于50 mL干燥二氯甲烷中,小心加入NaN3(2.60 g,40 mmol)搅拌使其溶解,0℃下将10 mL CF3COOH缓慢滴加到反应液中,反应1 h,室温反应4 h。TLC检测反应完成,0℃下,滴入NaHCO3饱和溶液至无气泡为止,分出有机层,无水Na2SO4干燥,减压蒸干,粗品用二氯甲烷:乙酸乙酯(1∶1)重结晶,得白色晶体4β-叠氮-4′-去甲基-4-脱氧表鬼臼毒素5 3.82 g,收率90%。将4β-叠氮-4′-去甲基-4-脱氧表鬼臼毒素5(4.25 g,10 mmol)溶于50 mL乙酸乙酯中,加入10%Pd/C(1.00 g),HCOONH4(2.52 g,40 mmol),加热回流搅拌5 h,TLC检测反应完全,过滤回收Pd/C,滤液用饱和食盐水洗3遍,减压蒸干得白色泡沫状固体粗产物4β-氨基-4′-去甲基-4-脱氧表鬼臼毒素6 3.63 g,收率88%,不经进一步分离纯化可直接用于下一步反应[10]。
1.3.2中间体3a-3c的合成将1 mmol糠醇1、1.2 mmol不同的二级胺和1.5 mmol甲醛水溶液溶于15 mL冰醋酸中,40℃反应2~4 h,TLC检测反应完全,减压尽量回收冰醋酸,然后用饱和NaOH溶液调节pH值为10-11,用乙酸乙酯萃取3次(3×30 mL),无水Na2SO4干燥,减压旋干后,溶于10 mL干燥CH2Cl2中,加入10 mmol活性MnO2室温反应2 h,减压旋干后,柱层析得到N-取代呋喃甲醛中间体3a-3c。
1.3.3目标化合物7a-7c的合成将0.6 mmol取代呋喃甲醛中间体3a-3c、4β-氨基-4′-去甲基-4-脱氧表鬼臼毒素6(0.5 mmol)溶于10 mL无水甲醇中,加入3滴冰乙酸做为催化剂,室温搅拌反应4~8 h,TLC检测反应完全。将反应液温度降至0℃,加入3 mmol NaBH4。加入完毕后,补加适量无水甲醇,0℃搅拌反应3~6 h,直至原料全部反应完毕(TLC检测)。滴加5%稀盐酸调节pH至7.5,减压旋干,加入20 mL蒸馏水超声使其呈混悬状态,加入等体积二氯甲烷萃取,有机相无水Na2SO4干燥,减压旋干得到粗品,粗品经柱层析(二氯甲烷:甲醇)纯化得到目标产物7a-7c,收率58%~75%。
1.3.4目标化合物11a和11b的合成将0.6 mmol取代呋喃甲醛中间体3a和3c、4β-氨基-4-脱氧表鬼臼毒素10(0.5 mmol)溶于10 mL无水甲醇中,加入3滴冰乙酸做为催化剂,室温搅拌反应4~8 h,TLC检测反应完全。将反应液温度降至0℃,加入3 mmol NaBH4。加入完毕后,补加适量无水甲醇,0℃搅拌反应3~6 h,直至原料全部反应完毕(TLC检测)。滴加5%稀盐酸调节pH至7.5,减压旋干,加入20 mL蒸馏水超声使其呈混悬状态,加入等体积二氯甲烷萃取,有机相无水Na2SO4干燥,减压旋干得到粗品,粗品经柱层析(二氯甲烷∶甲醇)纯化得到目标产物11a-11b,收率69%~71%。
1.4 MTT法检测体外抗肿瘤活性对数生长期细胞培养于96孔培养板内,每孔100 μL(含5 000~6 000个肿瘤细胞),置37℃,5%CO2温箱中培养。次日,给药组加入含有不同浓度的化合物,每种细胞设4~5个剂量组,每组至少设3个平行孔。对照组加入与给药组等体积的溶剂。置37℃,5%CO2温箱中培养。48 h后弃培养液,每孔加20μL 5 mg/mL MTT溶液(培养基配制)。37℃孵育4 h,弃上清液,每孔加入DMSO 150 μL溶解甲簪颗粒,轻度振荡溶解。用酶标仪在参考波长450 nm、检测波长570 nm条件下测定光密度值(OD),以溶剂对照处理的细胞为对照组,用下面公式计算药物对细胞的抑制率,根据计算得到的各浓度的抑制率通过Logit方法计算得到半数抑制浓度(I C50),重复测试3次,取平均值为最终结果[11]。

2 结果
2.1化合物的表征数据
7a:白色固体,收率:68%,mp:218-219℃;1HNMR(600 MHz,CDCl3)δ 6.45(s,1H,5-H),6.34(s,1H,8-H),6.27(s,2H,2′,6′-H),6.23(d,J=3.1 Hz,1H,4-Furan-H),6.21(d,J=3.1 Hz,1H,3-Furan-H),5.93(d,J=1.4 Hz,1H,α-OCH2O),5.91(d,J=1.4 Hz,1H,β-OCH2O),4.51(d,J=5.2 Hz,1H,1-H),4.32-4.19(m,2H,11-CH2-),3.91(d,J=3.9 Hz,1H,4-H),3.89(d,J=15.0 Hz,1H,α-CH2-Morpholine),3.76-3.73(m,4H,3,5-Morpholine-H),3.74(s,6H,3′,5′-OCH3),3.60(d,J=15.0 Hz,1H,β-CH2-Morpholine),3.57(d,J=1.9 Hz,2H,-CH2-Furan-),3.30(dd,J=13.8,5.3 Hz,1H,2-H),2.79-2.70(m,1H,3-H),2.55-2.49(m,4H,2,6-Morpholine-H).13C NMR(150 MHz,CDCl3)δ 175.51,153.36,147.63,147.25,146.39,133.99,132.38,131.82,131.13,110.09,108.54,108.37,108.00,101.29,68.45,66.73,56.43,55.44,54.63,53.36,46.61,43.52,41.36,38.62.HR-ESI-MS m/z:579.2352[M+H]+,理论值:579.2343[M+H]+。
7b:白色固体,收率:75%,mp:212-223℃;1H NMR(600 MHz,CDCl3)δ 7.31-7.27(m,2H,3,5-ArH),6.95(d,J=8.0 Hz,2H,2,6-ArH),6.89(t,J= 7.3 Hz,1H,4-ArH),6.48(s,1H,5-H),6.39(s,1H,8-H),6.31(s,1H,4-Furan-H),6.29(s,2H,2′,6′-H),6.25(d,J=3.0 Hz,1H,3-Furan-H),5.91(d,J=1.4 Hz,2H,-OCH2O),4.55(d,J=5.2 Hz,1H,1-H),4.35-4.25(m,2H,11-CH2-),3.95(d,J=3.8 Hz,1H,4-H),3.94(d,J=15.0 Hz,1H,α-CH2-Piperazine),3.78(s,6H,3′,5′-OCH3),3.70(s,2H,-CH2-Furan-),3.66(d,J =15.0 Hz,1H,β-CH2-Piperazine),3.34(dd,J=13.8,5.2 Hz,1H,2-H),3.32-3.26(m,4H,3,5-Piperazine-H),2.83-2.78(m,1H,3-H),2.78-2.68(m,4H,2,6-Piperazine-H).13C NMR(150 MHz,CDCl3)δ 175.46,151.09,149.60,147.65,147.24,146.39,146.30,133.93,132.32,131.82,131.14,129.12,129.10,119.90,116.17,116.14,110.11,108.62,108.35,108.03,107.97,101.25,68.43,68.01,67.60,56.48,56.44,54.72,52.74,48.77,46.64,43.52,41.35,38.61.HR-ESI-MS m/z:654.2818[M+H]+,理论值:654.2815[M+H]+。
7c:黄色固体,收率:58%,mp:218-219℃;1H NMR(600 MHz,CDCl3)δ 8.15(d,J=9.4 Hz,2H,3,5-ArH),6.85(d,J=9.4 Hz,2H,2,6-ArH),6.49(s,1H,5-H),6.44(s,1H,8-H),6.31(s,1H,4-Furan-H),6.30(s,2H,2′,6′-H),6.26(s,1H,3-Furan-H),5.93 -5.91(m,2H,-OCH2O),4.54(d,J=5.2 Hz,1H,1-H),4.34-4.22(m,2H,11-CH2-),3.95(d,J=3.8 Hz,1H,4-H),3.92(d,J=15.0 Hz,1H,α-CH2-Piperazine),3.79(s,6H,3′,5′-OCH3),3.72(d,J=15.0 Hz,1H,β-CH2-Piperazine),3.70-3.60(d,J=1.9 Hz,2H,-CH2-Furan),3.56-3.45(m,4H,3,5-Piperazine-H),3.33(dd,J=13.8,5.2 Hz,1H,2-H),2.83-2.76(m,1H,3-H),2.77-2.63(m,4H,2,6-Piperazine-H).13C NMR(150 MHz,CDCl3)δ 175.20,152.56,149.39,148.01,147.26,137.21,135.75,132.14,129.55,125.91,115.55,112.79,110.53,108.81,108.39,101.43,67.97,67.61,60.75,56.29,52.29,46.92, 44.09,41.44,37.69.HR-ESI-MS m/z:699.4907[M+ H]+,理论值:699.2666[M+H]+。
11a:白色固体,收率:69%,mp:219-220℃;1H NMR(600 MHz,CDCl3)δ 6.48(s,1H,5-H),6.37(s,1H,8-H),6.28(s,2H,2′,6′-H),6.27(s,1H,4-Furan-H),6.23(d,J=3.0 Hz,1H,3-Furan-H),5.96(d,J=1.3 Hz,1H,α-OCH2O),5.95(d,J=1.3 Hz,1H,β-OCH2O),4.55(d,J=5.3 Hz,1H,,1-H),4.35-4.24(m,2H,11-CH2-),3.94(d,J=3.4 Hz,1H,4-H),3.92(d,J=15.0 Hz,1H,α-CH2-Morpholine),3.80-3.75(m,4H,3,5-Morpholine-H),3.78(s,3H,4'-OCH3),3.76(s,6H,3′,5′-OCH3),3.66(d,J=15.0 Hz,1H,β-CH2-Morpholine),3.61(s,2H,-CH2-Furan-),3.35(dd,J=13.8,5.3 Hz,1H,2-H),2.83-2.75(m,1H,3-H),2.64-2.48(m,4H,2,6-Morpholine-H).13C NMR(150 MHz,CDCl3)δ 175.38,152.44,147.64,147.28,137.09,135.63,132.33,131.63,110.10,108.56,108.35,108.30,101.29,68.41,66.61,60.72,56.22,54.67,53.26,46.62,43.67,41.24,38.66.HRESI-MS m/z:593.2569[M+H]+,理论值:593.2499[M+H]+。
11b:黄色固体,收率:71%,mp:221-223℃;1H NMR(600 MHz,CDCl3)δ 8.13-8.09(m,2H,3,5-ArH),6.82-6.79(m,2H,2,6-ArH),6.45(s,1H,5-H),6.40(s,1H,8-H),6.25(s,2H,2′,6′-H),6.24(s,1H,4-Furan-H),6.21(d,J=3.0 Hz,1H,3-Furan-H),5.88(s,2H,-OCH2O),4.52(d,J=5.3 Hz,1H,1-H),4.31-4.21(m,2H,11-CH2-),3.91(d,J=3.9 Hz,1H,4-H),3.89(d,J=15.0 Hz,1H,α-CH2-Piperazine),3.79(s,3H,4′-OCH3),3.72(s,6H,3′,5′-OCH3),3.68(d,J=15.0 Hz,1H,β-CH2-Piperazine),3.64(s,2H,-CH2-Furan-),3.46(q,J=5.9,5.4 Hz,4H,3,5-Piperazine-H),3.31(dd,J=13.8,5.3 Hz,1H,2-H),2.80-2.73(m,1H,3-H),2.70-2.60(m,4H,2,6-Piperazine-H).13C NMR(150 MHz,CDCl3)δ 175.35,154.75,152.47,147.66,147.28,135.58,132.35,131.72,125.91,112.65,110.18,108.58,108.41,108.32,101.26,68.41,60.72,56.28,54.88,52.29,46.84,46.72,43.68,41.25,38.67.HR-ESI-MS m/z:713.2826[M+H]+,理论值:713.2823[M+H]+。
2.2细胞毒性测定测试5个新的鬼臼毒素衍生物的细胞毒活性(表1)。结果显示所合成的5个新化合物都具有细胞毒活性,其中化合物7c和11b对于耐药肿瘤细胞K562/A02的活性明显优于阳性对照依托伯苷。
表1 目标化合物对HeLa,K 562和K 562/A02细胞增殖的抑制活性(±s±s,n=3)Tab 1 Inhibiting activity of target com pounds against HeLa,K562and K 562/A02 proliferation(±s±s,n=3)
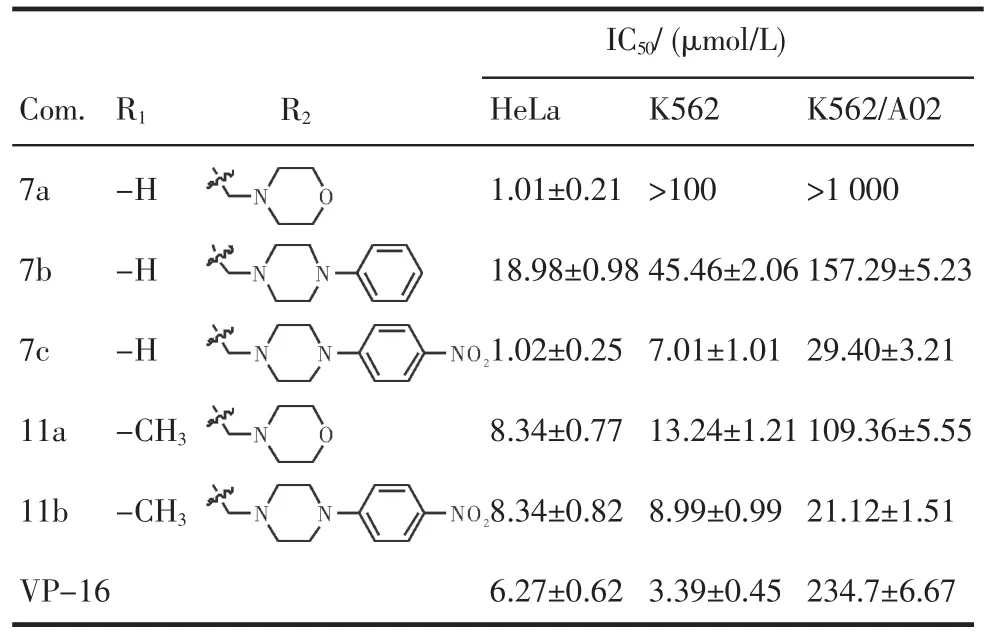
表1 目标化合物对HeLa,K 562和K 562/A02细胞增殖的抑制活性(±s±s,n=3)Tab 1 Inhibiting activity of target com pounds against HeLa,K562and K 562/A02 proliferation(±s±s,n=3)
IC50/(μmol/L)Com. 7a 7b 7c 11a 11b VP-16 R1 -H -H -H -CH3-CH3R2K562>100 45.46±2.06 7.01±1.01 13.24±1.21 8.99±0.99 3.39±0.45 K562/A02>1 000 157.29±5.23 29.40±3.21 109.36±5.55 21.12±1.51 234.7±6.67 HeLa 1.01±0.21 18.98±0.98 1.02±0.25 8.34±0.77 8.34±0.82 6.27±0.62
3 讨论
以天然产物鬼臼毒素结构为基础,设计并合成了5个未见文献报道的4β-N-取代呋喃鬼臼毒素衍生物,化合物结构经1H-NMR和13C-NMR和HRESI-MS确证,以依托泊苷为阳性对照,MTT法测试了所合成的5个4β-N-取代呋喃鬼臼毒素衍生物对HeLa,K562和K562/A02肿瘤细胞的细胞毒活性,结果表明,与阳性对照VP-16相比,所合成化合物细胞毒活性均较强,其中末端取代基为NO2的化合物,对于耐药的K562/A02细胞具有较强的活性,值得进一步研究。
[1]柳青.全球影响癌症发病的9种行为与环境危险因素[J].中华医学杂志,2006,86(1):44
[2]Imbert T F.Discovery of podophyllotoxins[J].Biochimie,1998,80(3):207
[3]Gordaliza M,García P A,del Corral J M,et al.Podophyllotoxin:distribution,sources,applications and new cytotoxic derivatives[J]. Toxicon,2004,44(4):441
[4]Hande K R.Etoposide:four decades of development of a topoisomerase II inhibitor[J].Eur J Cancer,1998,34(10):1514
[5]Xiao Z,Xiao Y D,Feng J,et al.Antitumor agents.213.Modeling of epipodophyllotoxin derivatives using variable selection k nearest neighbor QSAR method[J].J Med Chem,2002,45(11):2294
[6]Vanvliet D S,Tachibana Y,Bastow K F,et al.Antitumor agents. 207.Design,synthesis,and biological testing of 4beta-anilino-2-fluoro-4'-demethylpodophyllotoxin analogues as cytotoxic and antiviral agents[J].J Med Chem,2001,44(9):1422
[7]Zhou X M,Wang Z Q,Chang J Y,et al.Antitumor agents.120.New 4-substituted benzylamine and benzyl ether derivatives of 4'-O-demethylepipodophyllotoxin as potent inhibitors of human DNA topoisomerase II[J].J Med Chem,1991,34(12):3346
[8]Cheng W H,Cao B,Shang H,et al.Synthesis and evaluation of novel podophyllotoxin derivatives as potential antitumor agents[J].Eur J Med Chem,2014,85:498
[9]Cheng W H,Shang H,Niu C,et al.Synthesis and evaluation of new podophyllotoxin derivatives with in vitro anticancer activity[J]. Molecules,2015,20(7):12266
[10]Kamal A,Laxman N,Ramesh G.Facile and efficient one-pot synthesis of 4 beta-arylaminopodophy-llotoxins:synthesis of DNA topoisomerasellinhibitors(NPF and W-68)[J].Bioorg Med Chem Lett,2000,10(18):2059
[11]Jin J M,Zhang Y J,Yang C R.Spirostanol and furostanol glycosides from the fresh tubers of Polianthes tuberosa[J].J Nat Prod,2004,67(1):5
(2016-03-01收稿)
Synthesis and evaluation of furanoid podophyllotoxin derivatives w ith anticancer activity
CHENG Wei-hua1,2,NIU Cong3,SUN Qiang-wen4,CHEN Hong5,ZOU Zhong-mei2
(1.Institute of Medicinal Plant Development,Chinese Academy of Medical Sciences and Peking Union Medical College,Beijing 100193,China;2.Atom High Tech Co.LTD.,Beijing 102413,China;3.General Hospital of Chinese People's Armed Police Forces,Beijing 100039,China;4.8720 Hospital of Chinese People's Armed Police Forces,Wuxi 214000,China;5.Pharmacognosy Division,Medical College of Chinese People's Armed Police Force,Tianjin 300162,China)
O bjective:To acquire novel podophyllotoxin derivatives with higher antitumor activity.M ethods:The podophyllotoxin derivatives were synthesized using podophyllotoxin as the starting material through substituted furfuran and success split joint podophyllotoxin.Five 4β-N-substituted furfuran podophyllotoxin derivatives were designed,synthesized,and confirmed by1H-NMR,13C-NMR and HRMS,In vitro,cytotoxieities were tested against three human tumor cells(HeLa,K562,K562/A02)by MTT.Results:All compounds showed improved activities against HeLa and K562 when compared with VP-16.In particular,compound 7c,11b exhibited the most potent activity toward drug-resistant K562/A02 cells,as compared to VP-16.Conclusion:Substituted furan connected to the podophyllotoxin could increase antitumor activity.
podophyllotoxin;structure modification;antitumor activity.
R9
A
1006-8147(2016)05-0381-05
国家自然科学基金资助项目(30873363);国家“重大新药创制”科技重大专项(2011ZX09307-002-01 、2013ZX09508104)
成伟华(1985-),男,助理研究员,博士,研究方向:天然产物结构修饰及放射性药物研究;通信作者:邹忠梅,E-mail:zm zou@implad. ac.cn。
