格列美脲水凝胶剂体内外透皮释放的评价
2016-01-29杨文智李霞霞王树根李海鹰
杨文智,李霞霞,王树根,李海鹰
(河北大学 药学院,河北 保定 071002)
格列美脲水凝胶剂体内外透皮释放的评价
杨文智,李霞霞,王树根,李海鹰
(河北大学 药学院,河北 保定071002)
摘要:采用HPLC法检测自制格列美脲水凝胶的药物含量,研究凝胶中药物的体内外透皮释放.选用小鼠鼠皮,采用Franz立式扩散仪考察水凝胶中药物的体外释放,药物体外释放接近零级释放.选用新西兰兔背敷凝胶,测定兔体内格列美脲的药-时曲线.考察超声按摩兔皮后涂敷等量药物水凝胶,药物经皮渗透的变化.药代动力学研究结果表明:对照组血药质量浓度12 h达峰,ρmax为7.64 μg/mL,持续释药长达48 h.而兔皮超声后tmax提前至6 h,药物起效迅速.格列美脲水凝胶体内外释放呈相关性,即可通过体外释放状况预测药物体内吸收.
关键词:格列美脲;水凝胶;超声;体内外释放;透皮吸收
第一作者:杨文智(1972-),男,内蒙古锡林浩特人,河北大学副教授,从事生物医用材料及药物缓控释制剂方面研究.E-mail:wenzhi_yang@sina.com
格列美脲是FDA批准的第1个可联合胰岛素使用的口服降糖药,属第3代磺脲类药物,相比同类其他药物,具有起效快、作用持久及剂量小等优点[1],市售格列美脲剂型以片剂和胶囊为主.因为格列美脲水溶性差(1.6 μg/mL),溶出慢,影响其口服剂型发挥最佳临床疗效[2-3].另外,口服格列美脲剂型药物半衰期短,血药浓度波动大,需长时间用药,文献报道患者可出现低血糖症、胃肠道刺激和视觉障碍等不良反应[4-5],故改善口服格列美脲固体制剂溶出度的研究,时有报道[6].透皮给药制剂,可避免药物胃肠道反应和肝脏首过效应,维持平稳的血药浓度,制剂使用方便,易被患者接受.而格列美脲难溶,极大限制了其透皮给药新剂型的开发.查阅文献,未见格列美脲水凝胶剂的研究报道.基于此,本实验首先采用助溶技术解决格列美脲难溶的问题,选择卡波姆为凝胶基质,制备格列美脲水凝胶剂并考察凝胶剂中药物体内外释放,为格列美脲水凝胶处方设计及应用提供实验支持.
1仪器与试药
1.1 仪器
LC3000型高效液相色谱仪(北京创新通恒科技有限公司);Franz立式双室扩散仪(中科院上海有机所);B-629型超声波美容仪(15W,50-60Hz,吴邦美容美发设备有限公司);AN2651型分析天平(上海民桥精密科学仪器有限公司);HJ-6型磁力加热搅拌器(金坛市城东新瑞仪器厂);TGL-16G型离心机(上海安亭科学仪器厂);THZ-82型气浴恒温振荡箱(金坛市医疗仪器厂);KQ-250B型超声波清洗器(昆山市超声仪器有限公司);XH-C旋涡混合器(金坛市白塔新宝仪器厂);YQD-6B型氮吹仪(上海亭山仪表厂).
1.2 试剂
1.3 实验动物
昆明种小鼠(合格证号:1407010;18~22 g)和新西兰兔(合格证号:1407011;(2.5±0.1)kg),由河北省实验动物中心提供.
2方法与结果
2.1 格列美脲水凝胶剂的制备
精密称取格列美脲10 g和葡甲胺40 g,加去离子水50 mL,80 ℃水浴加热至完全溶解,搅拌放冷,得到20 mg/mL的药物溶液.搅拌条件下,按处方量依次加入氮酮(1%,体积分数)、油酸(1%,体积分数)、尼泊金乙酯(3 mg/mL)、EDTA-2Na(0.1 mg/mL)、甘油(10%,体积分数),混合均匀.然后加入卡波姆(质量浓度10 mg/mL),磁力搅拌,充分溶胀后加入三乙醇胺(质量浓度13.5 mg/mL),搅拌过夜,得到格列美脲水凝胶.
2.2 色谱条件的选择
2.2.1检测波长的确定精密称定格列美脲,配成适宜浓度的药物甲醇溶液,以甲醇为空白基质,用紫外可见分光光度计进行波长扫描.格列美脲的最大吸收波长为227 nm,且在此波长条件下,葡甲胺和空白基质均无干扰.
2.2.2色谱条件色谱柱:Hypersil BDS C18柱(46 mm × 250 mm,5 μm),分析柱前接ODS C18保护柱;流动相:甲醇-0.025 mol/L NH4H2PO4溶液(3∶1,体积比);流速:1.0 mL/min;检测波长:227 nm;柱温:常温;进样量:20 μL.
2.3 体外渗透实验
2.3.1实验方法采用Franz立式透皮扩散池,透过面积为2.69 cm2,接收池体积为13.0 mL.将处理的离体鼠皮固定于扩散池和接收池间,角质层朝上,确保真皮层与接受液紧密接触.以乙醇、聚乙二醇和生理盐水(5∶2∶3,体积比)混合液为接收液.精密称取格列美脲水凝胶4 g,含药60 mg,涂于小鼠皮肤表面,水浴温度(32 ± 0.5)℃,300 r/min搅拌速度,分别于0.5,1,2,4,6,8,10,12,24,36和48 h取出接收液,补充(32±0.5)℃等量接收液.取样5 000 r/min离心后,进样测定格列美脲含量.按下列公式分别计算格列美脲的单位面积累积释放量(Q)和药物透皮吸收百分率(W%),其中V为接收池的体积,ρn为第n次取样时的药物质量浓度,A为有效透过面积,M为水凝胶含药总量.

2.3.2标准曲线的建立精密称取恒重的格列美脲药物,用甲醇配成1 mg/mL的格列美脲储备液.稀释储备液配制成各浓度的工作液,按2.2.2方法进行分析,以峰面积A对药物浓度ρ(μg/mL)作图,得到格列美脲标准曲线为y= 5.8×104x + 3.5×104(R=0.9996;n=5),线性为0.1~100 μg/mL.紫外扫描显示,水凝胶基质对药物测定无干扰,格列美脲保留时间为7.8 min.

图1 格列美脲水凝胶剂中药物累积释放曲线
2.3.3精密度和回收率实验取一定浓度的格列美脲的对照品溶液,每隔2 h进行测定,连续测定5次,同一条件下,连续测定5 d,考察日内和日间精密度.日内RSD为0.20%,日间RSD为0.40%.在制备的凝胶剂中分别添加高(50 μg/mL)、中(5 μg/mL)、低(0.5 μg/mL)3种质量浓度的格列美脲标准液,3种质量浓度各5份,处理后分析测定.计算得到格列美脲的平均回收率为102%.以上结果表明:所建立的色谱法定量准确,方法可靠,可用于体外释放实验中格列美脲含量的测定.
2.3.4体外透皮实验结果根据公式分别计算格列美脲的单位面积累积渗透量(Q)和药物透皮吸收百分率(W%),并以Q对t作图,如图1所示.格列美脲在12 h内释放缓慢,12~48 h释放迅速,48 h格列美脲累积透过百分率达到处方量的27.2%.
Q~t回归分析,采用Higuchi方程、零级方程及一级方程进行拟合,选择相关系数(R2)最大的模拟方程为最优释放模型.结果见表1,水凝胶中格列美脲更倾向以零级动力学方程经皮渗透,属于Fick’s扩散渗透机制,水凝胶剂中格列美脲的累积释放量与时间呈良好的线性相关关系,R2为0.979 3,透皮速率常数J为124.39 μg/(cm2·h).

表1 格列美脲水凝胶剂中药物累积渗透量与时间的拟合方程
处方中以氮酮和油酸为混合促渗剂.文献报道,2种促渗剂稳定性好、对皮肤无毒无刺激、低浓度便可促进渗透[7].氮酮可改变角质层中脂质双分子层的致密性并增加其流动性,可减小药物在角质层中的扩散阻力,促进药物的透皮吸收[8].油酸属于顺式不饱和脂肪酸,广泛存在于自然界及人皮肤角质层中,作为皮肤内源性成分,具有良好的生物相容性[9].文献报道油酸与氮酮联用能发挥协同促透作用[10-11],故格列美脲水凝胶处方设计选用等量氮酮和油酸混合促透剂,以促进格列美脲透皮吸收.实验中采用离体小鼠鼠皮,尽量保持小鼠周龄、质量、皮肤选用部位及处理等因素相一致,减小个体差异对药物渗透的影响[12].
2.4 兔体内药代动力学实验
2.4.1动物分组和给药选取健康新西兰兔,雌雄各半,随机分为正常组和超声组,每组6只,实验前12 h禁食不禁水.去除兔背部体毛,保证皮肤无损伤,于正中至两侧划出5 cm×5 cm区涂抹给药,水凝胶均匀涂抹于兔背部其中一侧,给药剂量为18.36 mg/kg.正常组直接背敷水凝胶.超声组先用无水乙醇湿润兔背部皮肤,美容仪超声探头紧贴兔皮超声按摩25 min后,涂抹与正常组等量水凝胶.分别于0.5,1,2,4,6,8,12,24,36和48 h兔耳缘静脉取血0.5 mL,置于装有肝素钠的PE管中.采用DAS 2.0软件计算格列美脲在兔体内的药动学参数.
2.4.2血样的处理将加有肝素钠抗凝的全血5 000 r/min离心10 min,精密量取上清液20 μL,用1 mol/L的盐酸20 μL酸化10 min,加入混合有机溶剂(V(乙腈)∶V(二氯甲烷)=5∶1)1.2 mL,涡旋混合器5 min,8 000 r /min离心10 min,吸取上清液,50 ℃水浴,氮气吹干.残余物用50 μL甲醇溶解,12 000 r/min离心10 min,20 μL进样分析.
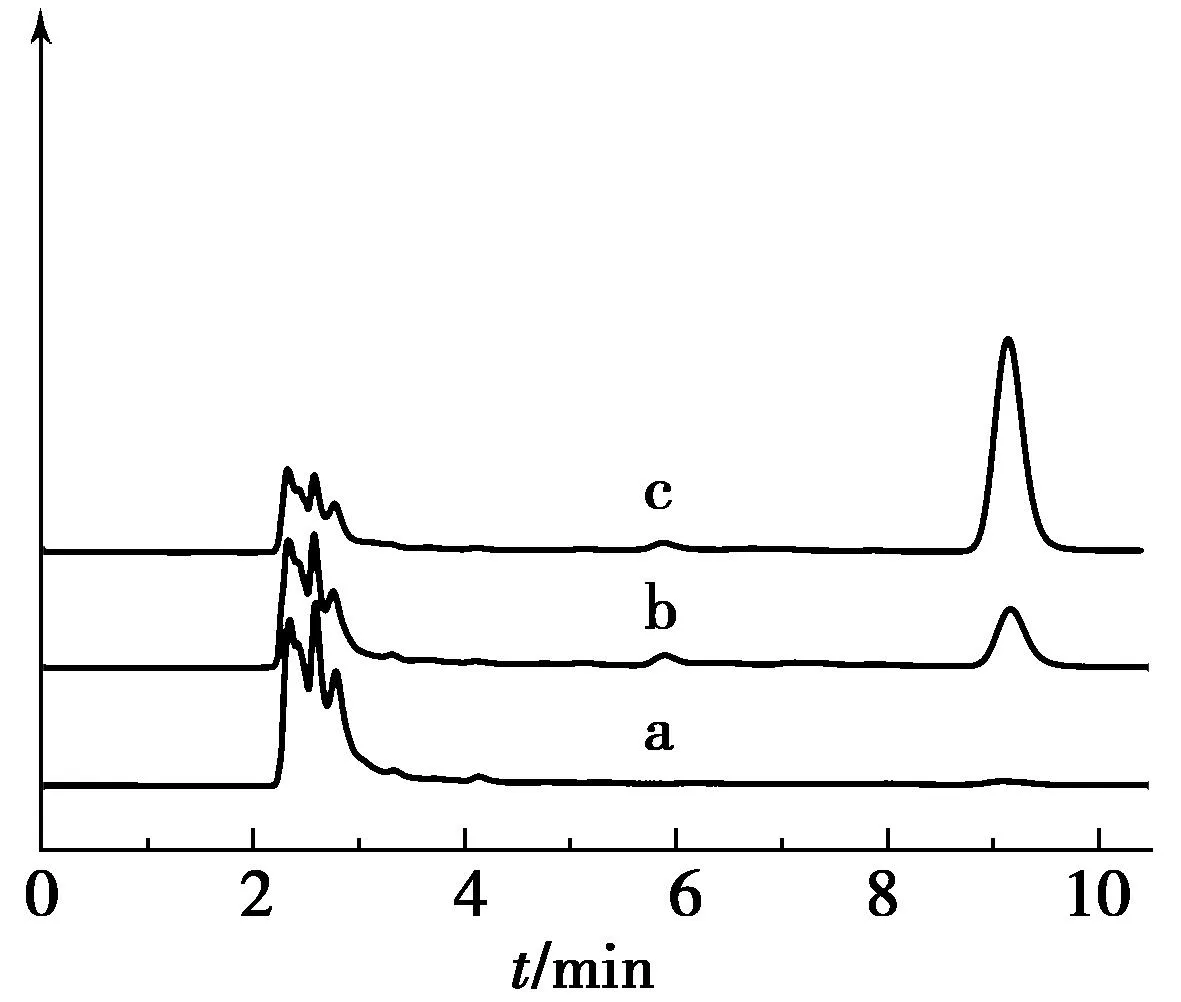
a.空白血浆;b.血浆样品;c.空白血浆加对照品.图2 格列美脲血样HPLC色谱Fig.2 HPLC spectrum

组别ρmax/(g·mL-1)Tmax/hAUC0-48/(g·h·mL-1)超声组7.616210.45正常组7.6412216.71

a.超声组;b.正常组.
2.5 体内外相关性
各时间点下格列美脲凝胶剂的兔体内药物累积AUC(Y)和体外单位面积药物累积渗透量(X)见表3.

表3 各时间点下格列美脲的体内外累积释放
以Y对X作图,见图4,得到回归方程Y= 0.039X+5.983,r=0.985.对相关系数r进行显著性检验,统计量t=r(n-2)1/2/(1-r2)1/2=17.12 >t0.001(10)=4.59,表明格列美脲水凝胶具备显著的动物体内外相关性,即通过测定格列美脲凝胶剂中药物体外释放情况可预测涂敷凝胶剂后药物在兔体内吸收状况,故获得实验数据可指导格列美脲水凝胶剂的临床应用.

图4 格列美脲体内累积AUC与体外单位面积累积渗透量的线性回归Fig.4 Linear regression plot of accumulated absorbed in vivo versus accumulated released in vitro
3结论
考察了格列美脲水凝胶剂中药物的体外释放和兔体内吸收,采用HPLC检测样品,分析方法可靠、准确.格列美脲水凝胶剂采用离体鼠皮的体外透皮实验显示,药物透皮接近零级释放,以恒定速率124.39 μg/(cm2·h)释放药物,48 h药物累积渗透约为处方量27%.兔体内药代动力学曲线表明,涂敷自制格列美脲水凝胶后,48 h内持续释放药物,具有缓释和长效作用.低频超声处理兔皮后涂敷等量格列美脲凝胶剂,显著缩短药物达峰时间,促进药物快速吸收.格列美脲水凝胶剂体内外释放呈现显著相关性,实验数据可为格列美脲水凝胶剂处方设计提供支持.
参考文献:
[1]AMMAR H O, SALAMA H A, GHORAB M, et al. Formulation and biological evaluation of glimepiride-cyclodextrin-polymer systems[J]. International Journal of Pharmaceutics, 2006, 309(1-2): 129-138.
[2]REVEN S, GRDADOLNIK J, KRISTL J, et al. Hyperbranched poly(esteramides) as solubility enhancers for poorly water-soluble drug glimepiride[J]. International Journal of Pharmaceutics, 2010, 396(1-2): 119-126.
[3]AMMAR H O, SALAMA H A, EL-NAHHAS S A, et al. Design and evaluation of chitosan films for transdermal delivery of glimepiride[J]. Current drug delivery, 2008, 5(4): 290-298.
[4]AHMED O A, AFOUNA M I, EL-SAY K M, et al. Optimization of self-nanoemulsifying systems for the enhancement of in vivo hypoglycemic efficacy of glimepiride transdermal patches[J]. Expert Opinion on Drug Delivery, 2014, 11(7): 1005-1013.
[5]韩刚,王成强,高远,等. 格列美脲壳聚糖贴剂的制备及经皮渗透研究[J]. 中国新药杂志,2011, 20(5): 456-458.
HAN Gang, WANG Chengqiang, GAO Yuan, et al. Preparation and transdermal penetration of glimepiride-chitosan patch in rats[J]. Chinese Journal of New Drugs, 2011, 20(5): 456-458.
[6]李海鹰,杨文智,马丽兰,等. 难溶药物格列美脲的片剂处方设计及溶出评价[J]. 河北大学学报:自然科学版,2014,34(5): 491-496.
LI Haiying, YANG Wenzhi, MA Lilan, et al. Preparation and dissolution rate evaluation of glimepiride tablets[J]. Journal of Hebei University Natural Science Edition, 2014, 34(5): 491-496.
[7]WILLIAMS A C, BARRY B W. Penetration enhancers[J]. Advanced Drug Delivery Reviews, 2012, 64(Suppl): 128-137.
[8]MAGHRABY G M M E, CAMPBELL M, FINNIN B C. Mechanisms of action of novel skin penetration enhancers: Phospholipid versus skin lipid liposomes[J]. International Journal of Pharmaceutics, 2005, 305(1-2): 90-104.
[9]TS M, VP D S, MB P. Influence of oleic acid on the rheology and in vitro release of lumiracoxib from poloxamer gels[J]. Journal of Pharmacy and Pharmaceutical Sciences, 2010, 13(2): 286-302.
[10]ABOOFAZELI R, ZIA H, NEEDHAM T E. Transdermal delivery of nicardipine: an approach to in vitro permeation enhancement[J]. Drug Delivery, 2002, 9(4): 239-247.
[11]张援,许东晖,许实波,等. 不同透皮吸收促进剂对格列美脲体外经皮渗透动力学特征的影响[J]. 中国药科大学学报, 2003, 34(6): 509-513.
ZHANG Yuan, XU Donghui, XU Shibo, et al. Effects of different penetration enhancers on the kinetics character of permeability of glimepiride through rabbit skin in vitro[J]. Journal of China Pharmaceutical University, 2003, 34(6): 509-513.
[12]李海鹰,杨文智,王苗苗,等. 水-氯涂膜剂中氯霉素体内外透皮释放评价[J]. 河北大学学报:自然科学版,2013, 33(5): 496-501.
LI Haiying, YANG Wenzhi, WANG Miaomiao, et al. Chloramphenicol release evaluation of salicylic acid and chloramphenicol plaster in vitro and in vivo[J]. Journal of Hebei University: Natural Science Edition, 2013, 33(5): 496-501.
[13]MITRAGOTRI S, BLANKSCHTEIN D, LANGER R. Transdermal drug delivery using low-frequency sonophoresis[J]. Pharmaceutical Research, 1996, 13(3): 411-420.
(责任编辑:梁俊红)

Drug release evaluation of glimepiride hydrogelinvitroandinvivo
YANG Wenzhi, LI Xiaxia, WANG Shugen, LI Haiying
(College of Pharmacy,Hebei University,Baoding 071002,China)
Abstract:In order to study and evaluate the release behavior of glimepiride(GM)hydrogel in vitro and in vivo, an HPLC method was used to determine the drug concentration in the hydrogel. The in vitro transdermal permeation behaviors of GM through mice skin were studied using Franz-type diffusion cell. The results showed that the drug released from hydrogel fitted with zero-order kinetic characteristics. A pharmacokinetic study of glimepiride hydrogel was carried out using New Zealand rabbits, and compared with the ultrasonic wave processing rabbit skin. It demonstrated that in the control group, the hydrogel could sustain release glimepiride for 48 h, and plasma drug mass concentration reached a peak(ρmax, 7.64 μg/mL) after 12 h. After an ultrasonic pretreatment, there was an earlier tmaxat 6 h.It was evident that ultrasonic could promote drug permeation. There was a good correlation between in vitro skin permeation and in vivo transdermal absorption using hydrogel, and the GM release in vitro could well predict its absorption in vivo.
Key words:glimepiride;hydrogel;ultrasonic;in vitro and in vivo;percutaneous absorption
基金项目:河北省自然科学基金资助项目(H2012201069;H2016201044);河北省科技计划项目(13272705)
收稿日期:2015-01-15
中图分类号:O658
文献标志码:A
文章编号:1000-1565(2015)06-0599-06
DOI:10.3969/j.issn.1000-1565.2015.06.008
