6个线粒体脑肌病伴高乳酸血症和卒中样发作综合征(MELAS)家系先证者线粒体基因全序列比较分析
2014-05-25刘莉邵宇权张宝荣蒋萍萍都爱莲管敏鑫
刘莉,邵宇权,张宝荣,蒋萍萍,都爱莲,4,管敏鑫
1. 浙江大学生命科学学院遗传学研究所,杭州 310058;
2. 浙江大学医学院附属邵逸夫医院神经内科,杭州 310016;
3. 浙江大学医学院附属第二医院神经内科,杭州 310009;
4. 上海交通大学医学院附属同仁医院神经内科,上海 200336
6个线粒体脑肌病伴高乳酸血症和卒中样发作综合征(MELAS)家系先证者线粒体基因全序列比较分析
刘莉1,邵宇权2,张宝荣3,蒋萍萍1,都爱莲3,4,管敏鑫1
1. 浙江大学生命科学学院遗传学研究所,杭州 310058;
2. 浙江大学医学院附属邵逸夫医院神经内科,杭州 310016;
3. 浙江大学医学院附属第二医院神经内科,杭州 310009;
4. 上海交通大学医学院附属同仁医院神经内科,上海 200336
线粒体脑肌病伴高乳酸血症和卒中样发作综合征(Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes, MELAS)是一种异质性很强的遗传代谢性疾病,而位于tRNALeu(UUR)基因的A3243G突变是该疾病最常见的致病位点。文章对6个汉族MELAS家系的先证者进行了临床病理、分子遗传学特征分析,探讨了线粒体基因多态性对MELAS病人表型可能产生的影响。线粒体基因检测结果显示,4例先证者为A3243G阳性,其异质性比例介于29%~59%之间,临床症状的严重性和异质性程度大致呈正相关;2例MELAS/Leigh叠加综合征先证者为A3243G阴性,复发次数和严重程度重于其他4例先证者,其中1例先证者的血液和肌肉组织中发现ND5基因T13094C突变,该位点已报道与MELAS/Leigh叠加综合征、小脑共济失调相关。另外,线粒体基因全序列测序结果显示:除主要致病突变外,还存在多个与耳聋、癫痫、糖尿病、心肌病、Leigh综合征相关的线粒体基因多态位点,临床症状严重的患者其多态位点也更多。这表明MELAS综合征的复杂表型不仅受致病突变位点的直接影响,也可能受到其他与疾病相关的多态性位点的修饰作用。
线粒体基因;线粒体脑肌病伴高乳酸血症和卒中样发作综合征;多态性;表型
线粒体脑肌病伴高乳酸血症和卒中样发作综合征(Mitochondrial encephalomyopathy with lactic acidosis and stroke-1ike episodes, MELAS, OMIM:540000)是一种临床和基因型高度异质性的线粒体病,常累及多个器官,以中枢神经系统、骨骼肌和心肌等高需能部位最为常见[1]。迄今为止,已报道与MELAS相关的突变位点多达40个以上(http://www.mitomap. org ),其中位于tRNALeu(UUR)基因的A3243G位点约占 80%[2]。然而 A3243G突变存在很大的临床异质性,使得携带同一个突变的个体呈现不同类型、不同程度的临床表型,有些甚至终身不发病。导致这种临床异质性的原因除了突变比例和组织分布的不同外,目前已报道有40余个mtDNA多态性位点也与MELAS表型有关[3],并且也有MELAS患者未携带A3243G的报道[4]。本文对6个MELAS家系的先证者进行了临床病理分析,并进行mtDNA全序列分析,探讨基因突变异质性及mtDNA多态性对患者表型的影响。
1 对象与方法
1.1 研究对象
6例临床诊断为MELAS的先证者(所属家系分别为HM1、HM2、HM3、HM4、HM5和HM6)和12例先证者家系母系成员(图 1)皆来自浙江大学医学院附属第二医院神经内科,研究内容和方法经浙江大学伦理委员会批准,并签署了知情同意书。所有先证者均进行血乳酸、神经电生理、头颅MRI和肌肉活检病理等检测。对家系成员的个人资料、亲属关系、家族史进行了详细的调查,并进行体格检查,以确定是否具有相关的临床表现。
1.2 方法
1.2.1 脑部影像学检查及肌肉病理学检测
对6例先证者在发病的急性期进行头颅MRI检查,包括T1加权、T2加权、扩散加权成像(DWI),其中 3例做了磁共振波谱分析(MRS)。肌肉病理:肌肉活检标本冰冻切片常规进行苏木精-伊红、改良Gomori三色、琥珀酸脱氢酶(SDH)及细胞色素c氧化酶(COX)染色等系列酶组织化学染色和透射电镜检测。
1.2.2 mtDNA基因检测与分析
抽取家系每个成员的外周血3 mL,苯酚-氯仿抽提法分离外周血全基因组DNA,-20℃保存备用,选取正常人DNA作为对照组。PCR扩增先证者家系若干母系成员及对照组的线粒体tRNALeu(UUR)基因,正向引物5′-TCAACAATAGGGTTTACGAC-3′ (mtDNA-2966-2985),反向引物5′-AGGAATGCCATTGCGATTAG-3′(mtDNA3346-3365),扩增产物用限制性内切酶ApaⅠ消化,3%琼脂糖凝胶电泳鉴定。野生型为 400 bp单一条带,突变型的 400 bp将被切成281 bp和119 bp两个片段。为进一步验证A3243G异质性程度与 MELAS临床表型及严重程度的相关性,采用焦磷酸测序法(BGI华大基因)精确测定A/G的突变比例。
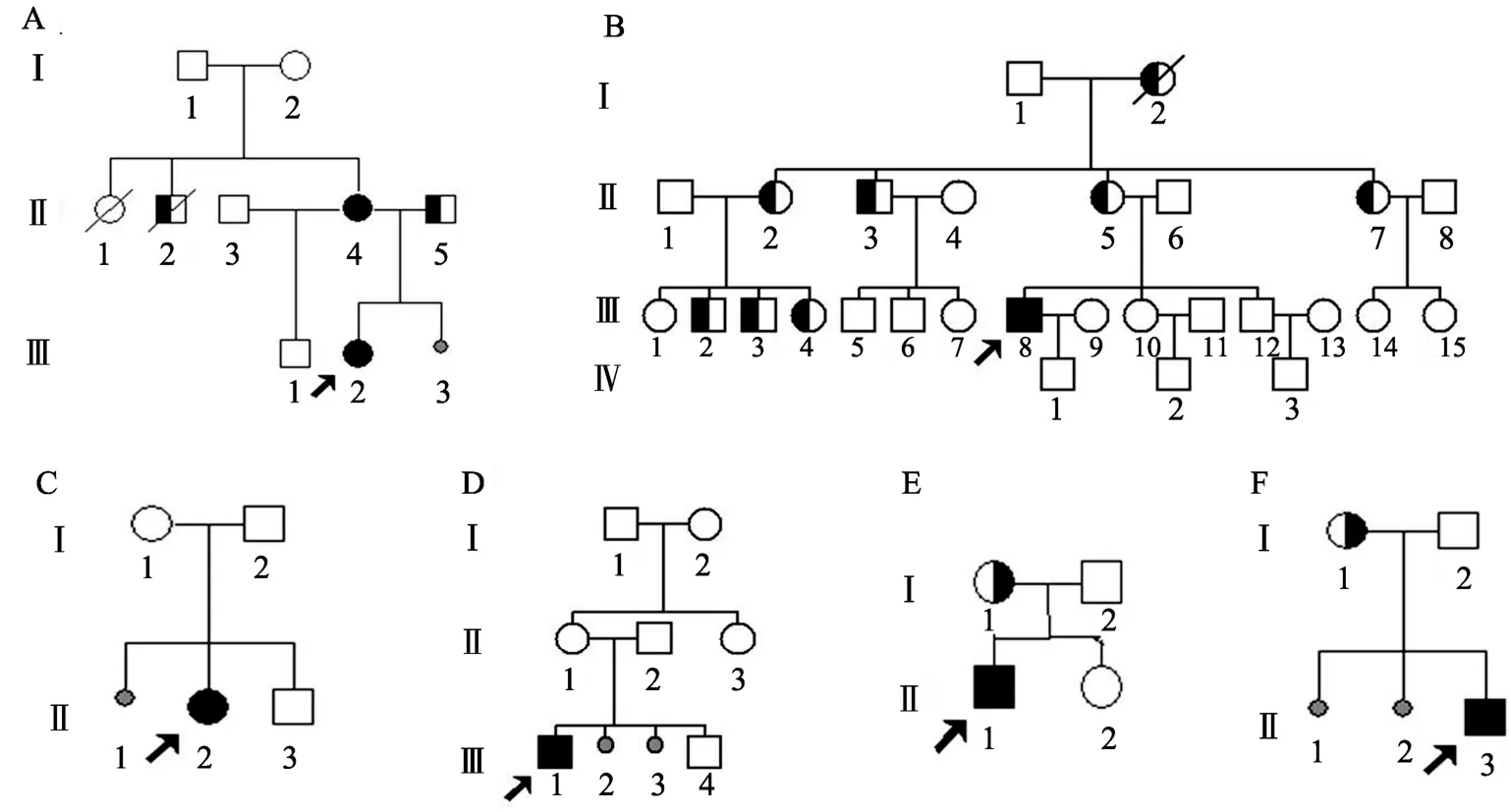
图1 6例MELAS先证者家系图A:HM1家系图;B:HM2家系图;C:HM3家系图;D:HM4家系图;E:HM5家系图;F:HM6家系图。表示耳聋患者;表示糖尿病患者;表示MELAS患者;表示正常人群,箭头表示先证者;表示流产。
线粒体基因全序列测定:使用24对具有部分片段重叠的引物[5],对先证者线粒体基因进行PCR扩增和Sanger测序,应用 SeqManII序列比对软件与修正后的人类线粒体标准序列[6]进行比对拼接,峰图查看分析软件为Chromas软件。为比较rRNA和编码区位点在进化上的保守性,查找人(NC_011137)、牛(HM045018.1)、鼠(NC_006914.1)、爪蟾(NC_ 001573.1)等 4个物种的 DNA及蛋白序列,采用BioEdit软件进行保守性分析。为探究多态性位点相关表型对 MELAS症状的修饰作用,根据线粒体全基因组测序情况采用Mitotool软件测定6例先证者的单倍型,并收集已报道的与线粒体病表型相关的多态性位点,比较 6例先证者多态性位点与表型的相关性。
对于血液中突变检测为阴性的患者,提取其肌肉组织DNA进行Sanger测序,以确认A3243G位点或其他可疑位点的致病性突变。
1.2.3 核基因POLG1和SURF1突变检测
POLG1和SURF1基因是与MELAS和Leigh综合征相关的常见核基因[7,8]。本研究扩增了两例A3243G为阴性的 MELAS/Leigh叠加综合征患者POLG1基因位于exon2的CAG重复序列,经1%琼脂糖凝胶电泳后进行直接测序;并对exon7、exon13、exon16、exon21区域进行了点突变检测。对SURF1基因进行了全基因测序。
2 结果与分析
2.1 MELAS家系临床资料评析
6例MELAS家系先证者临床资料显示(表1),先证者始发病年龄从13岁~36岁不等,临床表现均有反复的抽搐、脑卒中样发作和高乳酸血症等MELAS综合征典型症状;身材矮小、耳聋、认知退行、运动不耐受等常见伴发症状;本研究提示呼吸衰竭、消化道功能障碍、皮肤黑等症状也应值得关注。6例先证者影像学(图2A)均有典型的颞/顶枕大片异常信号,符合MELAS表现,3例先证者MRS有典型的乳酸波。2例(HM3和HM5)同时有中脑小脑异常信号,符合MELAS/Leigh重叠综合征表现,这2例先证者的复发次数明显多于其他4位先证者,其中HM3于发病后20个月死亡。肌肉活检(图2B)结果显示除HM5外,其他5例先证者肌肉中均见到破碎红纤维(Ragged red fibers, RRF)、线粒体数目和形态异常。血液乳酸含量检测发现,6例先证者中HM3的LAC含量最高(18 mmol/L),HM6的LAC含量最低(3.1 mmol/L),均高于正常人群(正常人<2 mmol/L)。
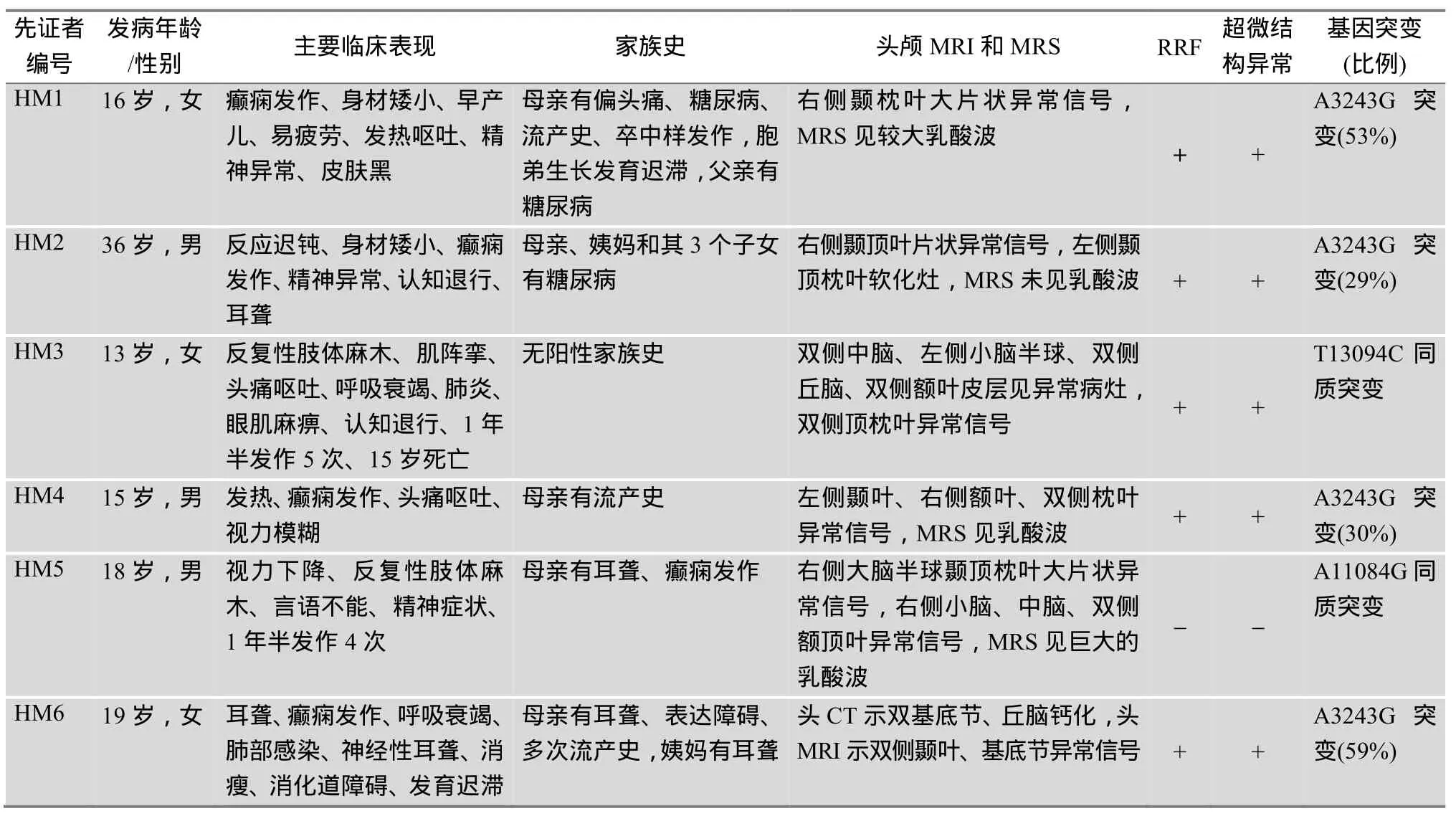
表1 6例MELAS家系先证者临床资料
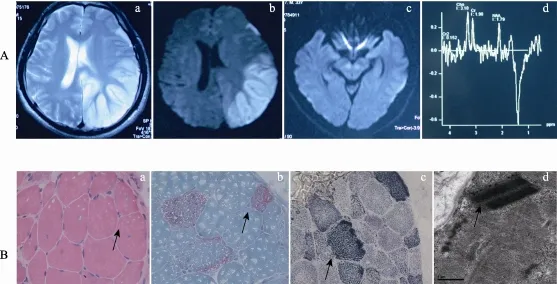
图2 先证者的头颅MRI和肌肉病理结果(以HM5-Ⅱ-2和HM6-Ⅱ-1为例)A:先证者HM5-Ⅱ-2的头颅MRI图像(a:T2相右侧颞枕叶大片状异常信号;b:DWI示右侧颞枕叶大片状异常信号弥散障碍;c:双侧中脑异常信号;d:MRS见1.33 ppm处巨大的乳酸波);B:先证者HM6-Ⅱ-1肌肉病理结果(a:HE染色示肌纤维大小不等,核内移;b:Gomori染色见破碎红纤维;c:SDH染色见周边强阳性纤维;d:电镜下见结晶样包涵体)。
结合6例先证者的家系图(图1),家族史中耳聋、糖尿病、流产史为常见的异常,其中HM1的母亲有偏头痛、糖尿病、流产史和卒中样发作 1次,为临床发病者;胞弟有生长发育迟滞,父亲有糖尿病,为高度异常家族史。HM2母亲、姨妈和其3个子女患有糖尿病。HM3无异常家族史,HM4母亲有流产史,HM5和HM6母亲均有耳聋表现。
2.2 线粒体基因组突变分析
2.2.1 A3243G突变位点检测及异质性分析
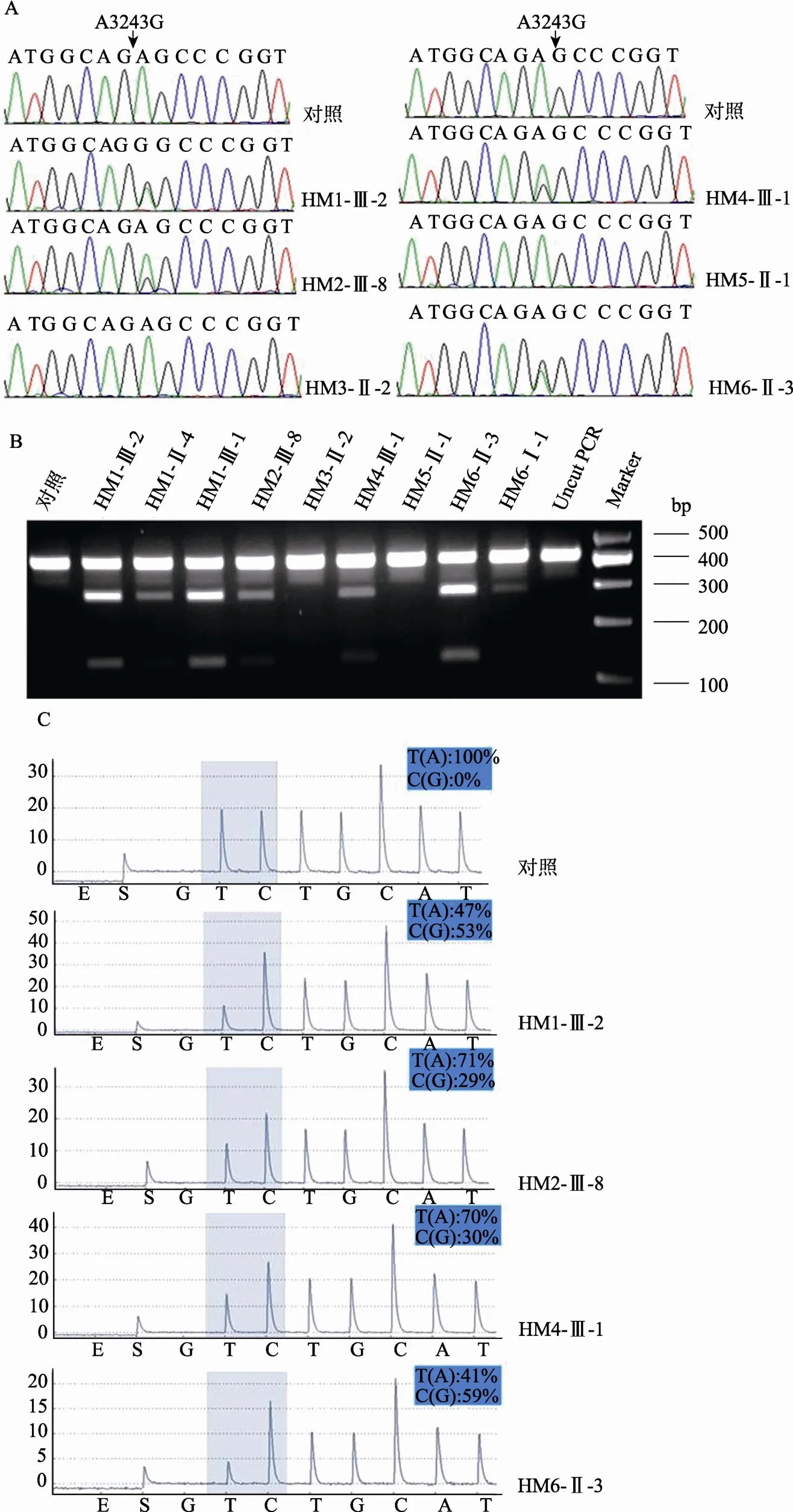
图3 A3243G突变检测和异质性分析A:A3243G位点Sanger测序结果;B:ApaⅠ酶切6例MELAS先证者A3243G电泳检测结果;C:4例A3243G阳性先证者焦磷酸测序异质性检测结果。
对6例先证者的tRNALeu(UUR)基因进行了Sanger测序。结果显示,4例先证者存在A3243G异质性突变且杂合峰高不同(图3A)。进一步对A3243G阳性先证者12位母系成员的基因组DNA进行PCR扩增,用ApaⅠ酶切鉴定,发现只有HM1的母亲(HM1-Ⅱ-4)和胞弟(HM1-Ⅲ-1)以及HM6的母亲(HM6-Ⅰ-1)存在A3243G异质性突变(图3B),而HM2家系的母系成员Ⅱ-5、Ⅲ-10、Ⅲ-12、Ⅳ-2和HM4家系的母系成员Ⅰ-2、Ⅱ-3、Ⅲ-4 中未检测到A3243G突变(数据未显示)。酶切结果显示(图3B):HM1和HM6先证者281 bp突变条带亮度明显高于HM2和HM4先证者。HM1和HM6先证者281 bp 条带亮度也高于其家系成员HM1-Ⅱ-4、HM1-Ⅲ-1和HM6-Ⅰ-1。为验证异质性存在的差异,对 A3243G阳性突变的先证者进行焦磷酸测序,结果显示异质性程度与酶切结果一致(图3C):HM1和HM6的异质性比例较高(图3C),达到53%和59%;而HM2和HM4的异质性分别为29%和30%。
2.2.2 A3243G阴性患者的突变位点分析
先证者HM3和HM5未检测到A3243G位点突变,也未发现与 MELAS相关的 C3256T、T3271C和T3291C等典型位点突变。但在HM3-Ⅱ-1血液中发现了T13094C突变,进一步检测其肌肉发现该位点突变在肌肉中的比例接近100%(图4)。对HM3的2位母系成员(Ⅰ-1、Ⅱ-3)的检测没有发现T13094C突变。已有报道T13094C突变可以导致MELAS/Leigh叠加综合征[9,10]。在 HM5患者中检测到 ND4基因A11084G同质性突变,该突变同时又是线粒体单体型A15a的多态位点,尚未见单独致病的报道。同时,本研究分析了HM3和HM5患者mtDNA中多态性位点。如表2所示,HM3和HM5中共有24个多态位点与线粒体疾病相关,其中HM3患者中有16个位点与MELAS相关,HM5中有10个位点与MELAS相关,同时与MELAS和Leigh综合征相关的位点有9个:D-Loop区A73G和A263G;12S rRNA A750G;16S rRNA A2706G;ATP6 A8701G;ND4 G11719A;ND5 C12705T和T13094C;Cytb A15326G。
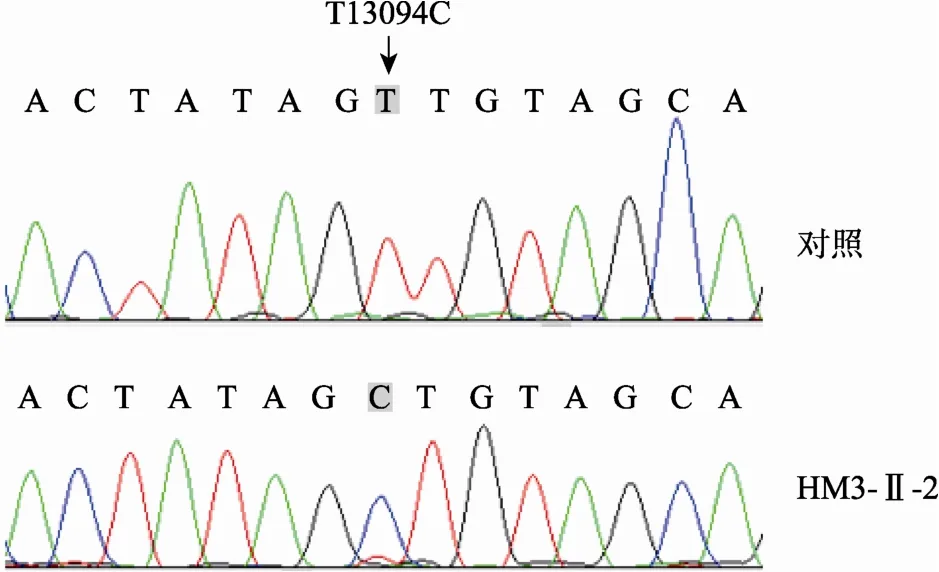
图4 HM3肌肉组织T13094C突变位点检测
2.2.3 mtDNA多态位点与临床症状比较分析
6例先证者共检测到已报道的多态性位点如下:D-loop区有13个;12S rRNA有6个;16S rRNA有3个;tRNALeu(UUR)基因有1个;在蛋白质编码区域存在10个已知的同义突变和22个已知的错义突变。这些错义突变包括:位于ND1的C3497T(A64V)、C3571T(L89F)、G4048A(D248N)、ND2 A4824G (T119A)、C5178A(L237M);CO2的A8108G(I175V);ATP8的C8414T(L17F)、A8459G(N32D); ATP6的G8584A(A20T)、T8618C(I31T)、A8701G(T59A)、C8794T(H90Y)、A8860G(T112A);ND3的A10398G (T114A);ND4的A11084G(T109A)、G11969A(A404T);ND5的T13094C(V253A);ND6的T14318C(N119S)、C14340T(V112M);Cyt b的C14766T(T7I)、T14979C (I78T)和A15326G(T194A)。这些突变的意义有待进一步研究。根据多态性位点的单倍体型分析,6例先证者的线粒体单倍体分别是:C5a1(HM1)、D4a (HM2和HM6)、M11(HM3)、B4c1(HM4)和A15a(HM5)。
MELAS患者往往是多器官受累,本研究对先证者线粒体基因组全序分析显示:无论是3243A>G突变阳性还是阴性患者,均存在多个与表型相关的多态性位点。这些突变位点大多与 MELAS、LHON、糖尿病、耳聋、偏头痛、心脏病、癫痫等症状相关。如D-Loop区域中,T146C与耳聋、LHON、线粒体肌病相关[12],C16278T与LHON、耳聋、代谢综合征、线粒体脑肌病相关[12];位于16S rRNA的G3010A和D-Loop的T16519C可促进伴随偏头痛的周期性呕吐综合征的发生;位于ND1区域的C3497T和位于ND4区域G11914A均与LHON表型相关。多态性位点T1095C、A8108G、T9950C、C14340T不仅与HM3先证者单倍体型位点组成相关,也与耳聋、LHON表型相关;同义突变位点 A750G、A1438G和A4769G与癫痫相关;同义突变位点G11719A和C12705T还与呼吸功能衰竭相关[3]。
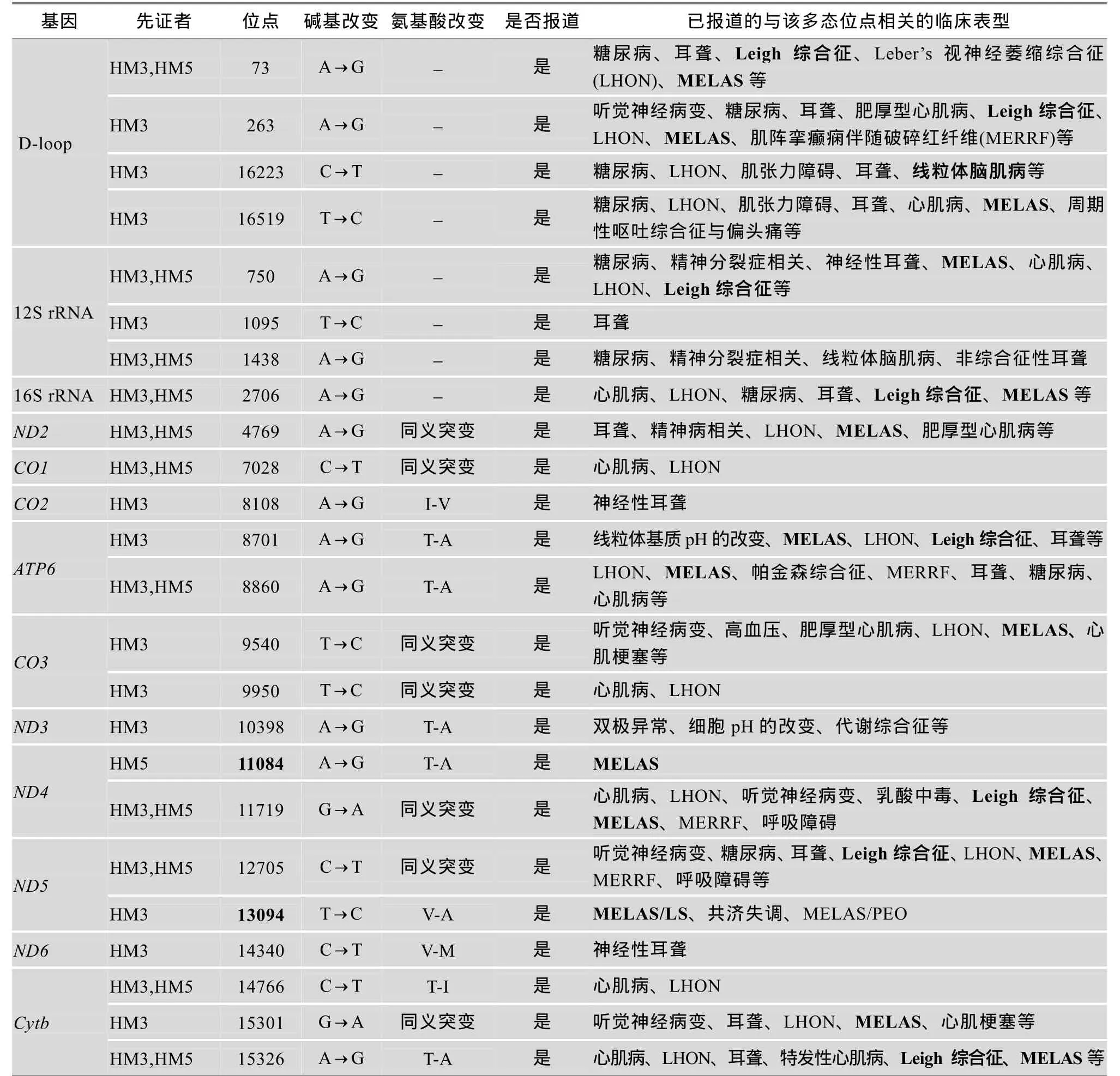
表2 2例A3243G突变阴性患者中与疾病相关的线粒体基因多态性位点
2.3 核基因POLG1和SURF1突变检测结果
对HM3-Ⅱ-1和HM5-Ⅱ-3患者的POLG1基因进行外显子2、7、13、16和21的PCR扩增,未找到与MELAS相关的典型突变位点,对SURF1基因测序分析比对也未发现致病性突变位点(exon1和2)。
3 讨 论
线粒体疾病是由于基因突变引起的、线粒体氧化磷酸化功能异常为特征的、多系统受累的疾病,无论是基因型还是临床表型均具有高度异质性。本研究探讨了6个MELAS家系先证者的临床表现和分子遗传学特征。家系中的先证者均表现出典型的MELAS临床症状,如头晕呕吐、精神异常、癫痫发作、认知退化、消瘦、身材矮小等;除 HM3和HM4家系成员无临床表现外,其他家系中的母系成员表现出了糖尿病、耳聋等症状,其中HM1先证者的母亲现在也是 MELAS患者。对先证者母系成员进行A3243G突变检测,发现HM1家系的Ⅱ-4、Ⅲ-1和HM6家系中的Ⅰ-1携带该突变,而HM2家系母系成员Ⅱ-5、Ⅲ-10、Ⅲ-12、Ⅳ-2和HM4家系的母系成员(Ⅰ-2、Ⅱ-3、Ⅲ-4) 中未检测到A3243G突变,这与国外报道的母系遗传方式[13]不相符。这一方面与突变比例低、检测灵敏度的限制有关,另一方面提示 A3243G也存在新生突变,即在卵细胞生成或者受精时期的自发突变[14]。
tRNALeu(UUR)A3243G 突变的杂合度往往与表型相关,但也有研究报道血液中杂合度与疾病严重程度并无明确的相关性[15]。A3243G突变与tRNALeu(UUR)的结构稳定性、氨酰化修饰、密码子的识别等有关[11]。该突变影响tRNA的合成并导致RNA转录剪切的终止,进一步使复合物Ⅰ和Ⅳ功能缺陷,从而导致ATP合成受阻,器官供能不足,导致临床症状的产生[16]。Lu等[17]研究提示当细胞中A3243G的杂合度超过70%不会引起糖尿病,而会出现更严重的症状,如身材矮小、原发性心肌病等;而当血液中杂合度在10%~30%时可能只引起糖尿病、耳聋等症状。本研究结果显示(图2A),阳性个体中HM1和HM6的杂合比例较高,HM2和HM4的杂合比例较低,除了HM4没有糖尿病或耳聋,HM1和HM2均有糖尿病,HM6有耳聋症状。Qi等[18]在57例A3243G突变的中国线粒体脑肌病患者中发现 A3243G异质性与表型的严重程度无关,但与发病年龄相关。Vivero等[19]则认为 A3243G异质性程度越高,耳聋表型就越严重,同时Ma等[20]在47例中国患者中探究到该异质性突变与疾病严重程度呈正相关。本研究中HM1和HM6两者异质性程度最高,典型的MELAS临床表现也较为严重,HM1的突变异质性高于其母亲Ⅱ-4,发病年龄也早于其母亲;HM6的母亲携带突变比例较低未发病,故本研究中的 4例 A3243G阳性患者表型严重程度与该位点异质性程度大致呈正相关。同时,本研究还发现突变的异质性程度越高,阳性家族史越突出,如 HM1家族中检出 2例A3243G突变者,其母亲又是临床发病者。
HM3和HM5虽然未检测到A3243G突变,但都具有典型的反复中风样发作、高乳酸血症、乳酸波等特征,MRI病变同时累及脑干和小脑符合MELAS/Leigh重叠综合征的影像学表现。本研究在HM3患者血液中检测到低比例的T13094C异质性突变,但肌肉组织中突变比率接近100%,已有研究报道该突变可以导致MELAS/Leigh叠加综合征、小脑性共济失调等表型[9,10],如此高的突变比例可以部分解释HM3患者频繁发作的代谢危象、日益恶化的脑功能乃至20个月内死亡的病程。线粒体多态位点分析发现该患者携带的多态性位点也最多,同时存在与耳聋、LHON、线粒体肌病、心肌病、头痛呕吐、呼吸衰竭和癫痫发作等临床表现相关的多个突变位点,其中2个位点(A10398G和G11719A)曾报道与乳酸中毒相关[3]。6例先证者皆有癫痫发作,除主要突变外,本研究在 6例患者中同时检测到了数个与癫痫相关的多态性位点,如 A750G、A1438G和A4769G[3]。由此我们推测在MELAS综合征中,临床表型的异质性不仅受致病突变位点的影响,也存在多态性位点的协同作用,从而使临床症状更复杂。这些多态性位点的协同作用使特定患者成为MELAS以及其他表型的易感人群,同时单倍型的不同可能从一定程度上解释不同家系之间表现出不同临床表型这一现象。
除线粒体基因外,有1500个核基因参与维持线粒体的正常生理功能[21]。SURF1基因突变是导致Leigh综合征的最常见基因,Cheldi等[7]认为在未找到mtDNA突变的不典型的MELAS患者,POLG1基因可以作为新的筛查指标,因此本研究对 2例MELAS/Leigh叠加综合征患者进行了 SURF1和POLG1基因检测,均未发现致病突变,但仍不能排除其他核基因参与致病的可能。值得一提的是:4例A3243G突变的患者经辅酶Q10、维生素B2、左卡尼汀治疗后,长时间处于稳定状态,而 2例A3243G突变阴性的MELAS/Leigh叠加综合征的患者,在同等药物干预下仍不断复发和进展,其中HM3于发病20个月内死亡,为线粒体脑肌病中急进类型,其发病机制很值得进一步探讨。
[1] Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol, 1984, 16(4): 481–488.
[2] Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M, Nonaka I. Mitochondrial myopathy, encephalopathy,lactic acidosis, and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology, 1992, 42(3): 545–550.
[3] MITOMAP: A Human Mitochondrial Genome Database. http://www.mitomap.org, 2014.
[4] Cevoli S, Pallotti F, La Morgia C, Valentino ML, Pierangeli G, Cortelli P, Baruzzi A, Montagna P, Carelli V. High frequency of migraine-only patients negative for the 3243 A>G tRNALeumtDNA mutation in two MELAS families. Cephalalgia, 2010, 30(8): 919–927.
[5] Rieder MJ, Taylor SL, Tobe VO, Nickerson DA. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res, 1998, 26(4): 967–973.
[6] Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet, 1999, 23(2): 147.
[7] Cheldi A, Ronchi D, Bordoni A, Bordo B, Lanfranconi S, Bellotti MG, Corti S, Lucchini V, Sciacco M, Moggio M, Baron P, Comi GP, Colombo A, Bersano A, on behalf of Lombardia GENS collaborators. POLG1 mutations and stroke like episodes: a distinct clinical entity rather than an atypical MELAS syndrome. BMC Neurol, 2013, 13: 8.
[8] Tiranti V, Hoertnagel K, Carrozzo R, Galimberti C, Munaro M, Granatiero M, Zelante L, Gasparini P, Marzella R, Rocchi M, Bayona-Bafaluy MP, Enriquez JA, Uziel G, Bertini E, Dionisi-Vici C, Franco B, Meitinger T, Zeviani M. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am J Hum Genet, 1998, 63(6): 1609–1621.
[9] Valente L, Piga D, Lamantea E, Carrara F, Uziel G, Cudia P, Zani A, Farina L, Morandi L, Mora M, Spinazzola A, Zeviani M, Tiranti V. Identification of novel mutations in five patients with mitochondrial encephalomyopathy. Biochim Biophys Acta, 2009, 1787(5): 491–501.
[10] Lax NZ, Hepplewhite PD, Reeve AK, Nesbitt V, McFarland R, Jaros E, Taylor RW, Turnbull DM. Cerebellar ataxia in patients with mitochondrial DNA disease: a molecular clinicopathological study. J Neuropathol Exp Neurol, 2012, 71(2): 148–161.
[11] Li RH, Guan MX. Human mitochondrial Leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR)A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and strokelike symptoms and diabetes. Mol Cell Biol, 2010, 30(9): 2147–2154.
[12] Chamkha I, Alila-Fersi O, Mkaouar-Rebai E, Aloulou H, Kifagi C, Hachicha M, Fakhfakh F. A novel m.12908T>A mutation in the mitochondrial ND5 gene in patient with infantile-onset Pompe disease. Biochem Biophys Res Commun, 2012, 429(1–2): 31–38.
[13] Servidei S. Mitochondrial encephalomyopathies: gene mutation. Neuromuscul Disord, 2004, 14(1): 107–116.
[14] Wang Z, Liu S, Yang Y, Yuan Y, Wu L, Qi Y, Chen Q. Detection of A3243G point mutation in mitochondrial DNA from 10 cases of MELAS. Chin Med J (Engl), 2002, 115(7): 995–997.
[15] Chinnery PF, Howell N, Lightowlers RN, Turnbull DM. Molecular pathology of MELAS and MERRF.the relationship between mutation load and clinical phenotypes. Brain, 1997, 120(10): 1713–1721.
[16] Hess JF, Parisi MA, Bennett JL, Clayton DA. Impairment of mitochondrial transcription termination by a point mutation associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature, 1991, 351(6323): 236–239.
[17] Lu JX, Wang DW, Li RH, Li WX, Ji JZ, Zhao J, Ye W, Yang L, Qian YP, Zhu Y, Guan MX. Maternally transmitted diabetes mellitus associated with the mitochondrial tRNALeu(UUR)A3243G mutation in a four-generation Han Chinese family. Biochem Biophys Res Commun, 2006, 348(1): 115–119.
[18] Qi Y, Zhang Y, Wang Z, Yang Y, Yuan Y, Niu S, Pei P, Wang S, Ma Y, Bu D, Zou L, Fang F, Xiao J, Sun F, Zhang Y, Wu Y, Wang S, Xiong H, Wu X. Screening of common mitochondrial mutations in Chinese patients with mitochondrial encephalomyopathies. Mitochondrion, 2007, 7(1–2): 147–150.
[19] Vivero RJ, Ouyang XM, Kim YG, Liu W, Du LL, Yan D, Liu XZ. Audiologic and genetic features of the A3243G mtDNA mutation. Genet Test Mol Biomarkers, 2013, 17(5): 383–389.
[20] Ma Y, Fang F, Cao Y, Yang Y, Zou L, Zhang Y, Wang S, Zhu S, Xu Y, Pei P, Qi Y. Clinical features of mitochondrial DNA m.3243A>G mutation in 47 Chinese families. J Neurol Sci, 2010, 291(1–2): 17–21.
[21] Wong LJC. Next generation molecular diagnosis of mitochondrial disorders. Mitochondrion, 2013, 13(4): 379–387.
(责任编委: 吴志英)
Mitochondrial genome analysis in the probands of six Chinese families with MELAS
Li Liu1, Yuquan Shao2, Baorong Zhang3, Pingping Jiang1, Ailian Du3,4, Minxin Guan1
1. Institute of Genetics, College of Life Science, Zhejiang University, Hangzhou 310058, China;
2. Department of Neurology, Sir Run Run Shao Hospital, School of Medicine, Zhejiang University, Hangzhou 310016, China;
3. Department of Neurology, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310009, China;
4. Tongren Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200336, China
Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS) is a geneticallyheterogeneous disorder. The most prevalent mitochondrial DNA (mtDNA) mutation associated with MELAS is the m.3243A>G transition in the mitochondrial tRNALeu(UUR)gene. Here, we report the clinical, genetic and molecular characterization of six probands from Han Chinese families with MELAS. Four of six probands carried the heteroplasmic m.3243A>G mutation. The levels of mutation load ranged from 29% to 59%, which were correlated with the severity of the clinical phenotypes. Two probands with MELAS/Leigh overlap were 3243 A>G negative, whose severity and relapse were greater than the other 4 probands. One proband with MELAS/Leigh harbored the known ND5 m.13094T>C mutation, which is related to MELAS/Leigh overlap and cerebella ataxia. Sequence analysis of entire mtDNA showed the distinct sets of variants including some variants that may be associated with diabetes, hearing loss, seizures, cardiomyopathy, and Leigh syndrome. Our data suggested that the phenotype and severity of MELAS mainly depend on the mutation load, and some variants may partially contribute to the phenotype and diversity. Our finding also highlighted the complexity of the relationship between genotype and phenotype in MELAS.
mitochondrial DNA; MELAS; polymorphism; phenotype
2014-04-04;
2014-06-03
国家自然科学基金项目(编号:81200967)资助
刘莉,硕士研究生,专业方向:遗传学。E-mail: 21207041@zju.edu.cn
都爱莲,副主任医师,硕士生导师,研究方向:线粒体脑肌病的基础和临床研究。E-mail: lotusdu@126.com
10.3724/SP.J.1005.2014.1159
时间: 2014-9-19 01:37:00 PM
URL: http://www.cnki.net/kcms/detail/11.1913.R.20140919.1337.002.html
