单细胞转录组高通量测序分析新进展
2014-05-25文路汤富酬
文路,汤富酬
北京大学生命科学学院生物动态光学成像中心,教育部细胞增殖与分化重点实验室,北京 100871
单细胞转录组高通量测序分析新进展
文路,汤富酬
北京大学生命科学学院生物动态光学成像中心,教育部细胞增殖与分化重点实验室,北京 100871
细胞异质性是生物组织的普遍特征。常规转录组测序(RNA-Seq)技术需要上万个细胞,所测结果实际上是一群细胞基因表达的平均值,所以难以鉴别细胞之间基因表达的异质性。单细胞 RNA-Seq技术的分辨率精确至单个细胞,为辨别异质性群体中各种细胞类型的转录组特征提供了有力的工具。近年来单细胞 RNA-Seq技术发展迅速,在方法学上包括cDNA扩增方法的多样化、对灵敏度和技术噪声的定量分析、浅覆盖高通量单细胞RNA-Seq方法和原位RNA-Seq技术等;在技术应用方面应用范围从早期胚胎发育扩大到组织器官发育、免疫和肿瘤等多个领域。文章对单细胞RNA-Seq在方法学和技术应用两方面的研究进展进行了详细阐述。
单细胞分析;转录组;高通量测序;RNA-Seq;异质性
自20世纪90年代以来,分子生物学技术的快速发展极大地提高了人们对转录组(Transcriptome)的分析能力,特别是随着高通量测序技术的发展,转录组测序(RNA-Seq)分析已经逐渐成为一种常用的实验手段[1]。然而在过去的20多年中,人们对转录组的认识仍然局限在群体细胞水平。这主要是由于技术的限制:常规转录组分析方法至少需要上万个细胞。通过这种群体细胞分析方法,所得到的实际上是一群细胞基因表达的平均值,主要反映该群细胞中数量占优势的细胞亚群的信息,而数量占劣势的细胞亚群的转录组特征则在平均化过程中被稀释。因此,常规转录组分析方法难以鉴别异质性群体中各种细胞亚群的转录组特征。但是,细胞异质性(Cell heterogeneity)却是生物系统和生物组织的普遍特征[2]。例如,在胚胎发育过程中,干细胞通过不断分裂分化产生各种特化细胞,其间还涉及许多过渡细胞类型;这些细胞类型互相交织镶嵌,具有重要生物学意义的细胞类型(如干细胞)经常只占少数。另外,看似同质的细胞之间基因表达也可存在很大差异,这部分源自基因表达内在的随机性(Stochastic)特征,可对细胞表型产生影响[3]。为了剖析这些细胞异质性,就有必要在单细胞水平进行基因表达分析。原位杂交和免疫组织化学方法能够提供单细胞分辨率的基因表达信息,但是只能同时检测少数基因。单细胞RNA-Seq技术能获得单个细胞内近万个基因的表达信息,为辨别生物组织中各种细胞类型的转录组特征和全面揭示细胞之间基因表达的异质性提供了有力的工具[4]。对单个细胞进行转录组分析,从 1990年 Brady等[5]报道的单细胞cDNA扩增方法开始,已有20多年的发展历史。2009年Tang等[6]首次报道了基于高通量测序的单细胞RNA-Seq技术。随着高通量测序的推广和发展,单细胞RNA-Seq技术近两年发展迅速。本文从方法学和技术应用两方面对单细胞RNA-Seq技术的最新研究进展进行阐述。
1 单细胞RNA-Seq方法学进展
单细胞 RNA-Seq与基于大量细胞的常规RNA-Seq相比,主要不同之处在于需要解决极微量起始样本量的问题。单个哺乳动物细胞内的总RNA含量约为10 pg,其中mRNA约为0.1~0.5 pg,而目前高通量测序文库需要至少数十纳克DNA,因此单细胞RNA-Seq的文库构建过程需将起始核酸进行数十万倍扩增[4]。如何避免核酸丢失和扩增偏差、提高技术的灵敏度和可重复性是单细胞RNA-Seq在方法学上首要解决的问题。另外,如何提高分析通量和节约成本,如何获得细胞的位置信息,也是亟待解决的问题。针对这些问题,本文从 4个方面阐述目前单细胞 RNA-Seq方法学的进展:(1)单细胞RNA-Seq的 cDNA扩增方法;(2)单细胞 RNA-Seq的灵敏度与技术噪声(Technical noise)分析;(3)浅覆盖(Low-coverage)高通量单细胞RNA-Seq方法;(4)原位RNA-Seq技术。
1.1 单细胞RNA-Seq的cDNA扩增方法
cDNA扩增方法是单细胞 RNA-Seq技术的核心,需尽可能完全地捕获起始样本中的目标RNA。一些常规 RNA-Seq方法如 Illumina TruSeq RNA Sample Prep试剂盒,在 cDNA扩增步骤之前将mRNA片段化,因而核酸丢失甚多。这些方法能够分析少至100 ng总RNA(约相当于1万个细胞),但难以对单个细胞进行分析。自Tang等[6]首次报道以来,近年又涌现出数种单细胞RNA-Seq的cDNA扩增方法,根据扩增步骤所采用的酶可分为PCR扩增(包括Brady/Tang方法[5,6]、Smart-Seq[7]、Smart-Seq2[8]、STRT-Seq[9]和SMA[10])、体外转录扩增(CEL-Seq[11])和Phi29聚合酶扩增(PMA[10])3类。其中,PCR扩增法根据第二链cDNA合成步骤所采用的方法又可分为末端加尾法、模板转换法和随机引物法。末端加尾法(Brady/Tang方法)采用末端脱氧核糖核酸转移酶在第一链cDNA 的3′末端加上一连串脱氧核苷酸(A或G),然后用多聚互补寡核苷酸(T或C)引物合成第二链 cDNA。模板转换法(包括 Smart-Seq、Smart-Seq2和STRT-Seq) 利用反转录酶的末端转移酶和模板转换活性合成第二链 cDNA。随机引物法(SMA)采用随机序列寡核苷酸引物合成第二链cDNA。这3种方法都在合成第二链cDNA的同时引入转录本5′端的锚定序列,从而与转录本 3′端的锚定序列(在合成第一链 cDNA时引入)成对,进而对cDNA进行PCR扩增。体外转录扩增法(CELSeq)不需要在合成第二链cDNA时引入锚定序列,其通过在合成第一链cDNA时引入RNA 聚合酶启动子序列,进而利用体外转录反应对cDNA进行扩增。这些方法更详细的介绍可参考本实验室最近的一篇综述文献[12],本文将着重讨论这些方法的灵敏度和技术噪声问题。
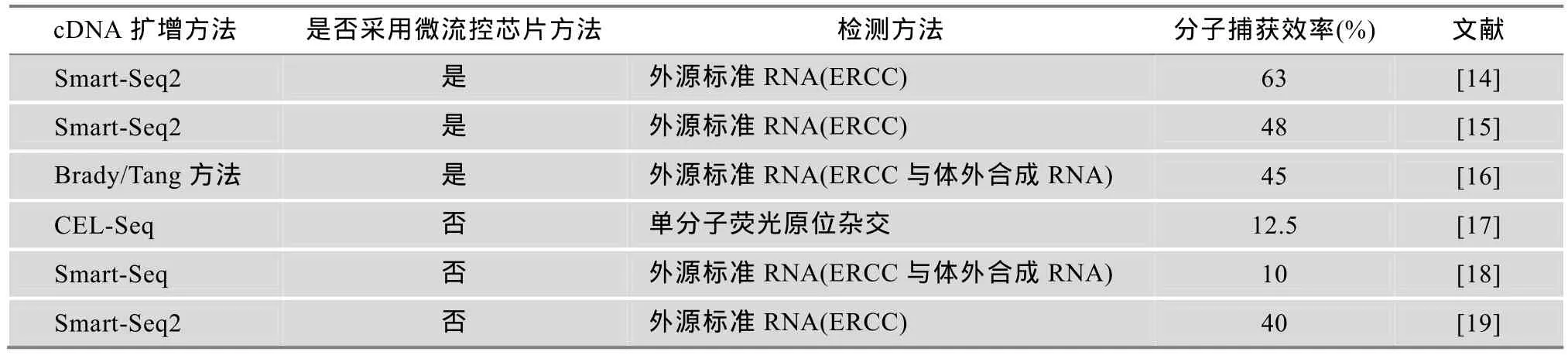
表1 单细胞RNA-Seq的分子捕获效率
1.2 单细胞RNA-Seq的灵敏度与技术噪声分析
灵敏度是指单细胞RNA-Seq捕获单个细胞内全部mRNA的比例。多个研究小组最近对其进行了定量分析[13~19]。在方法上主要通过掺入外源mRNA来判定:在单细胞裂解液中掺入已知数量的外源mRNA,比较外源mRNA所加入数量与所测得数量,即推断出单细胞 RNA-Seq的分子捕获效率(灵敏度)。总体而言,单细胞RNA-Seq的分子捕获效率为10%~63%。通过微流控芯片(Microfluidic)建库,效率要高于常规试管方法,这可能与微流控芯片方法能够减少mRNA丢失有关。3个研究小组采用微流控芯片方法报道的分子捕获效率为45%~63%[14~16],相比之下,另外 3个研究小组采用常规试管方法报道的分子捕获效率为 10%~40%[17~19](表 1)。另外,从cDNA扩增方法来看,Brady/Tang方法[16]与Smart-Seq2[14,19]接近,二者要高于Smart-Seq[18]与CEL-Seq[17](表1)。
单细胞 RNA-Seq的技术噪声是指单细胞RNA-Seq技术的系统误差所导致的测量值的波动,主要通过对同一样本或外源mRNA进行反复多次测量来判定。研究表明,单细胞RNA-Seq比大量细胞RNA-Seq的技术噪声大,低丰度基因比高丰度基因的技术噪声大。技术噪声主要来源于cDNA扩增以及文库构建过程每个反应步骤所产生的随机性:当起始mRNA含量减少到一定程度时,文库构建过程的随机性将出现指数放大,曲线基本符合泊松分布[13~17]。
如何提高单细胞RNA-Seq的灵敏度并降低其技术噪声呢?首先,单细胞RNA-Seq的灵敏度与技术噪声有内在的联系,通过优化反转录与PCR扩增反应效率能够同时提高灵敏度和减少技术噪声[8]。其次,采用独特分子识别(Unique molecular identifications)策略降低PCR扩增中产生的偏差,也能够减少部分技术噪声[15]。最后,通过掺入外源异种mRNA作为参照,可以判定技术噪声的边缘,从而能够更好地分辨出真实的信号[13]。
1.3 浅覆盖高通量单细胞RNA-Seq方法
体内单个细胞之间基因表达的异质性来源于多个方面,为了进行清晰地解析和全面地认识需要同时分析大量单个细胞。如何提高构建文库的通量,并降低测序成本,是单细胞RNA-Seq技术面临的实际问题。解决方案包括文献介绍的高通量单细胞定量PCR方法[12],以及本文具体介绍的两种浅覆盖高通量单细胞RNA-Seq方法[20,21]。
方法一:采用Fluidigm公司的C1单细胞自动制备系统,在微流控芯片中进行细胞裂解、反转录与cDNA扩增,并随后采用Illumina的Nextera试剂盒利用转座酶技术构建测序文库[14,20]。微流控芯片方法与转座酶技术显著提升了建库通量,通过该方法,研究者一次最多可以构建 96个单细胞的RNA-Seq测序文库。同时,研究表明,对每个细胞进行 5万条读数的浅测序就可以对细胞类型进行鉴定[20]。需要指出的是,该浅测序方法虽然能够区分谱系距离较远的细胞类型,如胚胎干细胞与神经细胞[20],但用于区分谱系距离近的细胞类型,如 I型与II型肺泡细胞,对每个细胞进行200~500万条读数的较浅测序应该更加可靠[22]。
方法二:采用CEL-Seq文库构建方法,结合条码法(Barcoding)实现高通量分析[11,21]。通过在反转录步骤给每个细胞加一段条码序列,随后将样品混合在一个试管中进行第二链cDNA合成、体外转录扩增和接头连接等步骤,该方法能够同时构建数百个单细胞的测序文库。因为只对mRNA分子的3′末端进行测序分析,所以每个细胞只需进行数万条读数的浅测序。
1.4 原位RNA-Seq技术
目前的单细胞RNA-Seq方法都需要将细胞从组织中分离和裂解以获取RNA,因而不可避免地丢失了细胞的位置信息。最近Church研究小组报道的荧光原位 RNA-Seq技术(Fluorescent in situ RNA sequencing, FISSEQ)向原位单细胞转录组分析迈出了重要一步[23]。该技术首先对固定于载玻片上的细胞进行反转录、cDNA环化与滚环扩增(Rolling-circle amplification, RCA)。在反应过程中掺入丙烯胺脱氧尿苷三磷酸并通过交联剂将扩增的cDNA固定于原位,从而使每个 RNA分子在原位形成直径为200~400 nm的DNA扩增产物球。随后通过Solid连接测序法对这些 DNA扩增产物球进行高通量测序分析。FISSEQ技术目前还存在许多尚待解决的问题,包括如何提高分子捕获效率和如何去除核糖体RNA等,但与组织切片结合的特征使该技术有望为生物学研究与医学临床诊断的单细胞转录组分析打开新的大门。
最近还报道了能同时检测多个目标基因的原位RNA检测技术[24~26]。其中包括:(1)将原位RNA锁式探针滚环扩增方法(Padlock-RCA)与 Solid连接测序法结合,报道能同时检测 39个目标基因[24];(2)在单分子荧光原位杂交(Single molecular fluorescent in situ hybridization, smFISH)技术的基础上进行荧光编码和多轮杂交,能同时检测12~32个目标基因[25,26]。虽然这些技术的基因检测数目尚未达到组学水平,但它们的高灵敏性和高成熟度使其在原位单细胞基因表达分析方面占有一席之地。
此外还要提到的是一种原位转录组分析(Transcriptome in vivo analysis, TIVA)方法,其可非侵袭性地分离活体环境中单个靶细胞的mRNA用于转录组分析[27]。该方法利用细胞穿膜肽将生物素多聚尿苷转运到活细胞内,后者能捕获mRNA,但被光敏反义寡核苷酸封闭。这样,尽管所有细胞内都有生物素多聚尿苷,只有被激光照射后的细胞,其mRNA才能被捕获。通过链霉亲和素磁珠收集这些mRNA即能进行后续的单细胞RNA-Seq分析。TIVA方法将有助于对一些复杂组织(如脑组织)进行活体单细胞转录组分析。
2 单细胞RNA-Seq技术应用进展
单细胞RNA-Seq技术的应用始于哺乳动物早期胚胎发育阶段的转录组分析[6,28,29],近两年随着技术的不断成熟,应用范围从早期胚胎发育[30,31]扩大到组织器官发育[20,22,32]、免疫系统[21,33,34]和肿瘤[7,35~37]等多个领域。除了这些研究报道,高通量单细胞定量PCR技术近年来也有不少优秀的研究工作[38~43]。
2.1 早期胚胎发育
哺乳动物早期胚胎发育阶段的细胞数量极为稀少,其转录组分析尤其需要单细胞RNA-Seq技术。近几年,小鼠[6,28,29,44]和人类[30,31]胚胎早期发育阶段的单细胞RNA-Seq分析被陆续报道。来自不同发育时期(卵细胞、受精卵、2-8细胞期、桑椹胚和囊胚)的单个胚胎细胞通过组成分分析(Principal component analysis)或无监督聚类(Unsupervised clustering)等生物信息学分析方法可以正确地归类,表明着床前胚胎细胞具有动态而规律的转录组变化[30,31]。细胞异质性在囊胚期变得明显,通过无监督聚类分析,囊胚细胞可自动聚类形成滋养外胚层、上胚层和原始内胚层3群细胞[30](图1A);在桑椹胚期,不同细胞群的标记基因则共表达于同一胚胎细胞中[38]。在囊胚期之前,胚胎细胞之间的细胞异质性尽管不显著,却已然显现。例如,在桑椹胚期,内层和外层细胞已可通过标记基因Sox2和Id2进行初步区分[38];甚至早在2至4细胞期,已有一些基因在卵裂球之间显示出差异表达[44]。
2.2 组织器官发育
组织器官发育是单细胞RNA-Seq技术应用的另一个重要领域。近年来陆续报道了多种组织器官(包括肺[22]、脑[20]、肾[32]、血液[41]和内耳[40])发育过程中的单细胞转录组分析。研究表明,单细胞RNA-Seq能够精细地分辨组织器官发育过程中包括过渡细胞类型在内的各种细胞类型的转录组。这种强大的分析能力充分体现于Quake研究小组对小鼠肺泡发育研究中[22]。小鼠的气道上皮细胞在 E16.5~E18.5 d开始分化,气道远端顶部形成囊泡,同时前体细胞分化为具有气体交换功能的I型肺泡上皮细胞(AT1)和能够分泌表面活性物质的 II型肺泡上皮细胞(AT2)。研究小组通过对E18.5 d小鼠胚胎肺远端组织的80个单细胞进行转录组分析,成功地鉴定出5个细胞亚群,其中包括 4个已知的分化细胞类型:AT1、AT2、纤毛上皮细胞和Clara细胞(图1B)。第5个细胞亚群表现出 AT1和 AT2共同前体细胞的特征,其不仅同时表达AT1和AT2二者的标记基因,并且在组成分分析中定位于二者之间(图 1B)。研究者发现了大量新的标记基因,包括 AT1中的 Hopx和 Vegfa、AT2中的 Egfl6和 Clara细胞中的 Krt15等。单细胞分辨率转录组图谱的绘制必将有力推动各种组织器官的发育生物学研究。
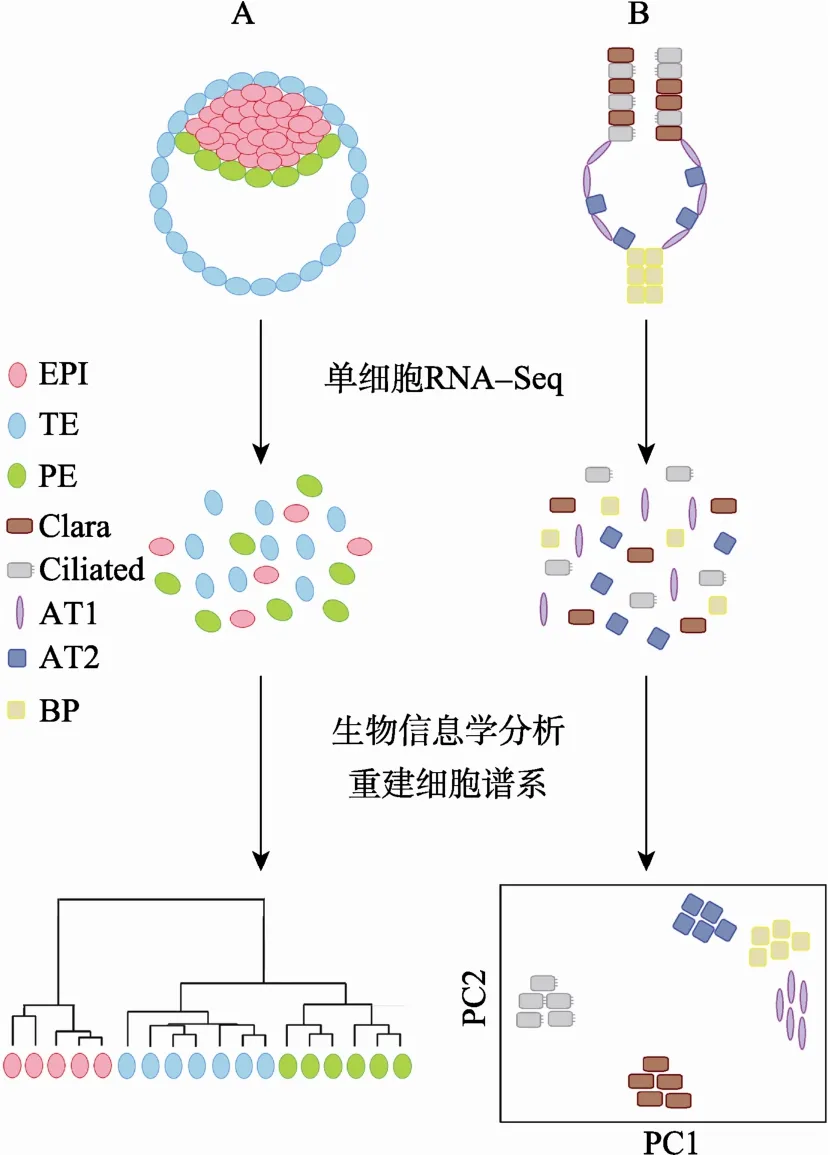
图1 单细胞RNA-Seq重建生物组织的细胞谱系囊胚早期胚胎发育(A)和肺泡组织器官发育(B)的单细胞RNA-Seq分析。将囊胚和肺泡组织分散为单个细胞,然后构建单细胞 RNA-Seq测序文库并进行高通量测序检测。采用包括无监督聚类(A)和组成分分析(B)在内的生物信息学方法进行数据分析,这些单细胞将自动聚类形成具有谱系结构的细胞群,每个细胞群对应一种细胞类型。这种单细胞RNA-Seq分析策略可以在单细胞水平重建生物组织的细胞谱系并获得其中各种细胞类型的转录组信息[22,30]。EPI:上胚层细胞;TE:滋养外胚层细胞;PE:原始内胚层细胞;Clara:Clara细胞;Ciliated:纤毛细胞;AT1:I型肺泡细胞;AT2:II型肺泡细胞;BP:双向分化潜能前体细胞。
2.3 免疫系统
随着高通量方法的发展,单细胞RNA-Seq技术近年来开始被应用于免疫系统和肿瘤的研究。免疫细胞对抗原物质的应答反应具有复杂和不均一性的异质性特点,单细胞RNA-Seq分析有望在此方面揭示前所未知的丰富信息。Shalek等[33,34]对脂多糖诱导的树突状细胞应答反应进行了系列的单细胞RNA-Seq研究:首先通过对18个树突状细胞进行分析,发现细胞成熟状态和基因表达的随机性特征是应答反应异质性的两个重要来源[33];随后采用浅覆盖高通量单细胞RNA-Seq对1700多个树突状细胞进行分析,进一步揭示出细胞应答反应的旁分泌机制[34]。Shalek等证明,树突状细胞群中的少部分细胞在病原体刺激早期(1~2 h)即出现一组所谓“核心”抗病毒基因的转录激活,这少部分细胞通过干扰素介导的旁分泌信号激活了该组基因在其余细胞中的表达;而另一方面,旁分泌机制在病原体刺激后期(2~4 h)又参与了对另一组炎症因子的转录抑制。Shalek等将单个树突状细胞隔离于微流控芯片小孔中,或者用药物抑制旁分泌,都能够抑制“核心”抗病毒基因组的转录激活和炎症因子组的转录抑制[34]。该研究所揭示的旁分泌机制可能是免疫系统增强病原体敏感性的一种有效策略。
2.4 肿瘤
细胞异质性是肿瘤的重要特征,可分为基因型和表型两个方面。基因型异质性是指同一肿瘤内部存在具有不同基因型的肿瘤细胞,而表型异质性是指同一肿瘤内部存在具有不同基因表达谱和功能特征的肿瘤细胞。最近报道的两篇肿瘤组织单细胞转录组分析充分展现了肿瘤细胞表型异质性的复杂性[35,39]。Dalerba等[39]通过单细胞定量 PCR技术发现结肠癌组织以及单个结肠癌干细胞形成的移植瘤组织中包含类似的多个分化细胞亚群,提示谱系分化是肿瘤细胞表型异质性的一个重要来源。Patel等[29]则对5例脑胶质母细胞瘤进行了数百个单细胞的RNA-Seq分析。他们的主要发现包括:(1)重要的肿瘤相关基因——如酪氨酸激酶受体家族基因(包括 EGFR和PDGFRA)——的表达水平和剪接形式在肿瘤细胞之间有明显差异,这种异质性可能是某些肿瘤对相应基因药物靶向治疗耐药的一个原因;(2)肿瘤细胞表型异质性可能部分来自瘤内微环境的不同,氧气和血供减少可能导致细胞休眠,进而可能产生化疗不敏感性;(3)体内瘤细胞呈现从类干细胞到分化细胞状态的连续变化,这与体外培养的瘤细胞明显不同,提示体外培养环境下的肿瘤干细胞模型只反映了某种特殊的细胞状态,而非体内肿瘤的全部细胞状态;(4)每个胶质母细胞瘤虽被归为某一亚型,但实际都以不同比例包含每种亚型的肿瘤细胞;(5)异质性高的肿瘤类型预后较差。Patel等的研究表明胶质母细胞瘤具有复杂的表型异质性。鉴于数目占少数的细胞亚群或许是肿瘤复发的源泉,肿瘤治疗可能需要关注所有的而不只是主要的细胞亚群。另外,最近还报道了黑色素瘤、前列腺癌和胰腺癌的循环肿瘤细胞(Circulating tumor cells, CTCs)的单细胞RNASeq分析[7,36,37]。循环肿瘤细胞是一种从肿瘤原发灶中脱落进入患者血液的肿瘤细胞,数量极为稀少,但与肿瘤的转移和复发有着密切关系[45]。单细胞RNASeq为其转录组分析提供了工具。研究表明,循环肿瘤细胞的转录组与原发瘤接近,但是一些与 Wnt信号通路[37]、细胞周期和癌症抗原有关的基因表达上调。研究者还发现了候选的生物标记物,有助于开发新的循环肿瘤细胞捕获方法[7]。
2.5 其他
最后,单细胞RNA-Seq还被用于揭示更为基础的细胞异质性现象:单等位基因转录现象[19]。双倍体细胞的每个基因座位上都有两个等位基因;在群体细胞水平,通常两个等位基因都被检测到转录。最近 Deng等[19]发现,在单细胞水平,存在着一种普遍的、动态和随机的单等位基因转录现象。在每个单细胞中(包括胚胎干细胞和成熟细胞),大约12%~24%的常染色体基因是单等位基因转录的。鉴于这种转录方式呈现出一种动态和随机的特点,其可能来于基因表达的随机性特征。
3 结语与展望
近年来,单细胞RNA-Seq技术在多个方面迅速发展。首先,多种cDNA扩增方法的涌现和对灵敏度和技术噪声的定量分析使其在技术类型和理论上更加完善;其次,浅覆盖高通量单细胞RNA-Seq方法的发展和商品化为其广泛应用提供了良好基础;再次,原位RNA-Seq技术将单细胞转录组分析指向了更广阔的应用领域;最后,在早期胚胎发育、组织器官发育、免疫和肿瘤生物学方面的实践应用充分展示了该技术鉴定异质性群体中各种细胞类型转录组的强大分析能力(图1)。
随着方法学的成熟,单细胞RNA-Seq技术在未来数年中将会得到更为广泛的应用。可能包括如下几个方面:(1)对各种组织器官的发育过程进行单细胞转录组分析,发现新的细胞类型和标记基因;(2)对神经和免疫系统进行全面分析,揭示其细胞异质性;(3)分析各种类型的肿瘤,揭示其细胞异质性;(4)与迅速发展的基因组编辑技术结合,对复杂的转录调控网络进行单细胞分辨率分析[46]。单细胞RNA-Seq技术将逐渐应用到医学临床实践之中,对人类早期胚胎细胞[30,31]和循环肿瘤细胞[7,36]的开创性工作已经指向了这种可能性。
[1] Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet, 2009, 10(1): 57–63.
[2] Kalisky T, Blainey P, Quake SR. Genomic analysis at the single-cell level. Annu Rev Genet, 2011, 45: 431–445.
[3] Huang S. Non-genetic heterogeneity of cells in development: more than just noise. Development, 2009, 136(23): 3853–3862.
[4] Tang F, Lao K, Surani MA. Development and applications of single-cell transcriptome analysis. Nat Methods, 2011, 8(Suppl.4): S6–S11.
[5] Brady G, Barbara M, Iscove NN. Representative in vitro cDNA amplification from individual hemopoietic cells and colonies. Methods Mol Cell Biol, 1990, 2: 17–25.
[6] Tang FC, Barbacioru C, Wang YZ, Nordman E, Lee C, Xu NL, Wang XH, Bodeau J, Tuch BB, Siddiqui A, Lao KQ, Surani MA. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods, 2009, 6(5): 377–382.
[7] Ramsköld D, Luo SJ, Wang YC, Li RB, Deng QL, Faridani OR, Daniels GA, Khrebtukova I, Loring JF, Laurent LC, Schroth GP, Sandberg R. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol, 2012, 30(8): 777–782.
[8] Picelli S, Björklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods, 2013, 10(11): 1096–1098.
[9] Islam S, Kjällquist U, Moliner A, Zajac P, Fan JB, Lönnerberg P, Linnarsson S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res, 2011, 21(7): 1160–1167.
[10] Pan XH, Durrett RE, Zhu HY, Tanaka Y, Li YM, Zi XY, Marjani SL, Euskirchen G, Ma C, LaMotte RH, Park IH, Snyder MP, Mason CE, Weissman SM. Two methods for full-length RNA sequencing for low quantities of cells and single cells. Proc Natl Acad Sci USA, 2013, 110(2): 594–599. [11] Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep, 2012, 2(3): 666–673.
[12] 文路, 汤富酬. 单细胞转录组分析研究进展. 生命科学, 2014, 26(3): 228–233.
[13] Brennecke P, Anders S, Kim JK, Kołodziejczyk AA, Zhang XW, Proserpio V, Baying B, Benes V, Teichmann SA, Marioni JC, Heisler MG. Accounting for technical noise in single-cell RNA-seq experiments. Nat Methods, 2013, 10(11): 1093–1095.
[14] Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, Mburu1 FM, Mantalas GL, Sim S, Clarke MF, Quake SR. Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods, 2014, 11(1): 41–46.
[15] Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lönnerberg P, Linnarsson S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods, 2014, 11(2): 163–166.
[16] Streets AM, Zhang XN, Cao C, Pang YH, Wu XL, Xiong L, Yang L, Fu YS, Zhao L, Tang FC, Huang YY. Microfluidic single-cell whole-transcriptome sequencing. Proc Natl Acad Sci USA, 2014, 111(19): 7048–7053.
[17] Grün D, Kester L, van Oudenaarden A. Validation of noise models for single-cell transcriptomics. Nat Methods, 2014, 11(6): 637–640.
[18] Marinov GK, Williams BA, McCue K, Schroth GP, Gertz J, Myers RM, Wold BJ. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res, 2014, 24(3): 496–510.
[19] Deng QL, Ramsköld D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science, 2014, 343(6167): 193–196.
[20] Pollen AA, Nowakowski TJ, Shuga J, Wang XH, Leyrat AA, Lui JH, Li NZ, Szpankowski L, Fowler B, Chen PL, Ramalingam N, Sun G, Thu M, Norris M, Lebofsky R, Toppani D, Kemp DW, Wong M, Clerkson B, Jones BN, Wu SQ, Knutsson L, Alvarado B, Wang J, Weaver LS, May AP, Jones RC, Unger MA, Kriegstein AR, West JA. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol, 2014, 32(10): 1053–1058.
[21] Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, Amit I. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science, 2014, 343(6172): 776–779.
[22] Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, Quake SR. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature, 2014, 509(7500): 371–375.
[23] Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, Terry R, Jeanty SSF, Li C, Amamoto R, Peters DT, Turczyk BM, Marblestone AH, Inverso SA, Bernard A, Mali P, Rios X, Aach J, Church GM. Highly multiplexed subcellular RNA sequencing in situ. Science, 2014, 343(6177): 1360–1363.
[24] Ke RQ, Mignardi M, Pacureanu A, Svedlund J, Botling J, Wählby C, Nilsson M. In situ sequencing for RNA analysis in preserved tissue and cells. Nat Methods, 2013, 10(9): 857–860.
[25] Lubeck E, Cai L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat Methods, 2012, 9(7): 743–748.
[26] Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L. Single-cell in situ RNA profiling by sequential hybridization. Nat Methods, 2014, 11(4): 360–361.
[27] Lovatt D, Ruble BK, Lee J, Dueck H, Kim TK, Fisher S, Francis C, Spaethling JM, Wolf JA, Grady MS, Ulyanova AV, Yeldel SB, Griepenburg JC, Buckley TP, Kim J, Sul JY, Dmochowski IJ, Eberwine J. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat Methods, 2014, 11(2): 190–196.
[28] Tang FC, Barbacioru C, Bao SQ, Lee C, Nordman E, Wang XH, Lao KQ, Surani MA. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell, 2010,6(5): 468–478.
[29] Tang FC, Barbacioru C, Nordman E, Bao SQ, Lee C, Wang XH, Tuch BB, Heard E, Lao KQ, Surani MA.Deterministic and stochastic allele specific gene expression in single mouse blastomeres. PLoS ONE, 2011, 6(6): e21208.
[30] Yan LY, Yang MY, Guo HS, Yang L, Wu J, Li R, Liu P, Lian Y, Zheng XY, Yan J, Huang J, Li M, Wu XL, Wen L, Lao KQ, Li RQ, Qiao J, Tang FC. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol, 2013, 20(9): 1131–1139.
[31] Xue ZG, Huang K, Cai CC, Cai LB, Jiang CY, Feng Y, Liu ZS, Zeng Q, Cheng LM, Sun YE, Liu JY, Horvath S, Fan GP. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature, 2013, 500(7464): 593–597.
[32] Brunskill EW, Park JS, Chung E, Chen F, Magella B, Potter SS. Single cell dissection of early kidney development: multilineage priming. Development, 2014, 141(15): 3093–3101.
[33] Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu DN, Trombetta JJ, Gennert D, Gnirke A, Goren A, Hacohen N, Levin JZ, Park H, Regev A. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature, 2013, 498(7453): 236–240.
[34] Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, Lu DN, Chen PL, Gertner RS, Gaublomme JT, Yosef N, Schwartz S, Fowler B, Weaver S, Wang J, Wang XH, Ding RH, Raychowdhury R, Friedman N, Hacohen N, Park H, May AP, Regev A. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature, 2014, 510(7505): 363–369.
[35] Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suvà ML, Regev A, Bernstein BE. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science, 2014, 344(6190): 1396–1401.
[36] Cann GM, Gulzar ZG, Cooper S, Li RB, Luo SJ, Tat M, Stuart S, Schroth G, Srinivas S, Ronaghi M, Brooks JD, Talasaz AH. mRNA-Seq of single prostate cancer circulating tumor cells reveals recapitulation of gene expression and pathways found in prostate cancer. PLoS ONE, 2012, 7(11): e49144.
[37] Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, Ciciliano JC, Smas ME, Winokur D, Gilman AJ, Ulman1 MJ, Xega K, Contino K, Alagesan B, Brannigan BW, Milos PM, Ryan DP, Sequist LV, Bardeesy N, Ramaswamy S, Toner M, Maheswaran S, Haber DA. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature, 2012, 487(7408): 510–513.
[38] Guo GJ, Huss M, Tong GQ, Wang CY, Li SL, Clarke ND, Robson P. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev Cell, 2010, 18(4): 675–685.
[39] Dalerba P, Kalisky T, Sahoo D, Rajendran PS, Rothenberg ME, Leyrat AA, Sim S, Okamoto J, Johnston DM, Qian DL, Zabala M, Bueno J, Neff NF, Wang JB, Shelton AA, Visser B, Hisamori S, Shimono Y, van de Wetering M, Clevers H, Clarke MF, Quake SR. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol, 2011, 29(12): 1120–1127.
[40] Durruthy-Durruthy R, Gottlieb A, Hartman BH, Waldhaus J, Laske RD, Altman R, Heller S. Reconstruction of the mouse otocyst and early neuroblast lineage at single-cell resolution. Cell, 2014, 157(4): 964–978.
[41] Guo GJ, Luc S, Marco E, Lin TW, Peng C, Kerenyi MA, Beyaz S, Kim W, Xu J, Das PP, Neff T, Zou KY, Yuan GC, Orkin SH. Mapping cellular hierarchy by single-cell analysis of the cell surface repertoire. Cell Stem Cell, 2013, 13(4): 492–505.
[42] Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, Klemm SL, van Oudenaarden A, Jaenisch R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell, 2012, 150(6): 1209–1222.
[43] McDavid A, Dennis L, Danaher P, Finak G, Krouse M, Wang A, Webster P, Beechem J, Gottardo R. Modeling bi-modality improves characterization of cell cycle on gene expression in single cells. PLoS Comput Biol, 2014, 10(7): e1003696.
[44] Biase F, Cao XY, Zhong S. Cell fate inclination within 2-cell and 4-cell mouse embryos revealed by single-cell RNA sequencing. Genome Res, 2014, doi: 10.1101/gr. 177725.114. [Epub ahead of print]
[45] Alix-Panabières C, Schwarzenbach H, Pantel K. Circulating tumor cells and circulating tumor DNA. Annu Rev Med, 2012, 63: 199–215.
[46] Wen L, Tang FC. Reconstructing complex tissues from single-cell analyses. Cell, 2014, 157(4): 771–773.
(责任编委:方向东)
Recent progress in single-cell RNA-Seq analysis
Lu Wen, Fuchou Tang
Biodynamic Optical Imaging Center, College of Life Sciences, Ministry of Education Key Laboratory of Cell Proliferation and Differentiation, Peking University, Beijing 100871, China
Cell heterogeneity is a general feature of biological tissues. Standard transcriptome analysis approaches require tens of thousands of cells to provide an average view of gene expression and ignore the information of gene expression heterogeneity. The single-cell RNA-Seq technologies profile gene expression at the single-cell level and serve as powerful tools to identify distinct phenotypic cell types within a heterogeneous population. The single-cell RNA-Seq technologies have been developed rapidly in recent years. The methodological progress includes a variety of cDNA amplification methods, the quantitative analysis of the sensitivity and noise of the technologies, and the development of the low-coverage high-throughput single-cell RNA-Seq and the in situ RNA-Seq technologies. Furthermore, the scope of application is extended from early embryonic development to tissue and organ development, immunology and oncology. In this review, we discuss recent progress in methodology and applications of the single-cell RNA-Seq technologies.
single-cell analysis; transcriptome; high-throughput sequencing; RNA-Seq; cell heterogeneity
2014-08-27;
2014-09-17
国家自然科学基金项目(编号:31271543)资助
文路,博士,助理研究员,研究方向:单细胞分析,基因组学。E-mail: wenlu.wl@gmail.com
10.3724/SP.J.1005.2014.1069
时间: 2014-10-8 10:02:13
URL: http://www.cnki.net/kcms/detail/11.1913.R.20141008.1002.001.html
