QuEChERS结合液相色谱质谱联用技术测定动物源性食品中N-二甲基亚硝胺
2024-04-15赵广西刘志梅
赵广西 刘志梅


摘 要:目的:建立高效液相色谱质谱联用技术定量检测动物源性食品中的N-二甲基亚硝胺。方法:样品前处理粉碎后经乙腈提取,低温离心除去油脂,QuEChERS净化,采用多反应监测模式测定。结果:N-二甲基亚硝胺校准曲线在2.0~200.0 ng·mL-1线性关系良好,相关系数为0.998 5,方法检出限和定量限分别为0.5 μg·kg-1和3.0 μg·kg-1,回收率为86.8%~90.8%,相对标准偏差在2.2%~3.1%。结论:该方法取样量少,试剂消耗量少,提取和净化流程简便,定性和定量准确度好,可用于检测动源性食品中N-二甲基亚硝胺。
关键词:高效液相色谱-质谱联用技术;QuEChERS;N-二甲基亚硝胺;多重反应监测
Determination of N-Dimethylnitrosamine in Food of Animal Origin by QuEChERS Combined with LC-MS/MS
ZHAO Guangxi1, LIU Zhimei2
(1.Qinhuangdao Municipal Food and Drug Inspection Center, Qinhuangdao 066000, China; 2.Qinhuangdao Administration of Market Regulation, Qinhuangdao 066000, China)
Abstract: Objective: To establish a high performance liquid chromatography-mass spectrometry method for the quantitative determination of N-dimethylnitrosamine in animal derived food. Method: The sample was extracted by acetonitrile after pretreatment and grinding, and the oil was removed by low temperature centrifugation, purified by QuEChERS, and determined by multi-reaction monitoring mode. Result: The calibration curve of N-dimethylnitrosamine had a good linear relationship in the range of 2.0~200.0 ng·mL-1, and the correlation coefficient was 0.998 5. The detection limit and quantitative limit of the method were 0.5 μg·kg-1 and 3.0 μg·kg-1, respectively. The recovery rate was 86.8%~90.8%, and the relative standard deviation was 2.2%~3.1%. Conclusion: This method has the advantages of less sampling, less reagent consumption, simple extraction and purification process, good qualitative and quantitative accuracy, and can be used to detect N-dimethylnitrosamine in animal-derived food.
Keywords: high performance liquid chromatography-mass spectrometry; QuEChERS; N-dimethylnitrosamine; multiple reaction monitoring
肉、魚等食品中富含蛋白质和脂肪,在煎、炸、烤等热处理过程中会产生较多的胺类化合物,易与亚硝酸盐反应生成亚硝胺类化合物,其中主要是N-亚硝基二甲胺。N-亚硝胺类化合物具有基因毒性,能够直接或间接损伤细胞DNA。实验证明,该类化合物有强致癌性,小剂量长期接触或较大剂量冲击都可引发癌症[1-4]。国际癌症研究机构在1987年将亚硝胺列为2A级致癌物。多国对食品中N-亚硝胺类化合物限量进行了严格的规定,美国农业部规定食品中总挥发性亚硝胺限量为10 μg·kg-1;我国《食品安全国家标准 食品中污染物限量》(GB 2762—2022)规定,肉制品中N-二甲基亚硝胺限量为3.0 μg·kg-1,水产品中限量为4.0 μg·kg-1[5-6]。
亚硝胺在食品中含量极微,一般在μg·kg-1水平,且检测过程中易受复杂背景基质的干扰。前处理是整个分析检测过程的关键和难点环节,影响分析的各项指标。样品前处理技术主要有固相萃取、固相微萃取、凝胶渗透色谱以及QuEChERS等技术[7-8]。亚硝胺检测方法主要有气相色谱-热能分析联用法、气相色谱-串联质谱法和液相色谱-串联质谱法。其中,液相色谱-串联质谱法因具有高灵敏度和普适性较好的特点,被广泛应用于食品中亚硝胺的检测[9-10]。
本研究建立了一种基于大气压化学电离源的高效液相色谱-质谱联用法测定动物源性食品中N-二甲基亚硝胺的方法,该方法操作简单、灵敏度高,可用于普通实验室对不同动物源性食品中亚硝胺的快速检测,为食品安全提供实际参考依据。
1 材料与方法
1.1 仪器与试剂
LC 20A液相色谱仪(岛津);AB5500三重四极杆质谱(AB);氮吹仪(安谱);高速冷冻离心机(Sigma);高速万能粉碎机(泰斯特)。
甲醇(色谱纯,TEDIA);乙腈(色谱纯,TEDIA);甲酸(色谱纯,Sigma);实验用水为超纯水(Millipore);N-二甲基亞硝胺标准液(CAS:62-75-9,浓度1 000 μg·mL-1,坛墨质检-标准物质中心)。
1.2 样品前处理
称取5 g(精确到0.01 g)干制品于50 mL离心管中(鲜样品称取10 g,精确到0.01g),加入10 mL水,振荡10 min,加入乙腈5 mL,涡旋振荡3 min,于-18 ℃冷冻60 min,加入5 g硫酸镁和2 g氯化钠,涡旋振荡2 min,2 ℃、9 000 r·min-1冷冻离心10 min,移取上清液。
称取150 mg HLB,100 mg无水硫酸镁粉末于15 mL离心管中,加入上清液涡旋振荡2 min,置于冷冻离心机,9 000 r·min-1、2 ℃离心5 min。取2 mL上清液,转入试管中,控制氮吹温度在15 ℃低速氮吹至0.9 mL,准确定容至1.0 mL,0.25 μm滤膜过滤,进行检测。
1.3 色谱/质谱条件
色谱条件:Waters UPLC BEH C18色谱柱(150 mm×2.1 mm,1.7 μm)分离,以0.1%甲酸的水溶液(A)、0.1%甲酸的甲醇溶液(B)进行梯度洗脱;流速:0.4 mL·min-1;柱温:40 ℃;多重反应监测模式下正离子模式定量分析。梯度洗脱程序:0.0~3.0 min,10%B;3.0~6.0 min,95%B;6.0~7.0 min,95%B;7.0~9.0 min,10%B;9.0~10.0 min,10%B。具体质谱条件见表1。
1.4 标准工作溶液配制
准确移取N-二甲基亚硝胺标准品溶液100 μL于10 mL棕色容量瓶中,用二氯甲烷定容,得到浓度为10 μg·mL-1的标准溶液;再次用二氯甲烷稀释成浓度分别为2 ng·mL-1、5 ng·mL-1、10 ng·mL-1、20 ng·mL-1、50 ng·mL-1及200 ng·mL-1的N-二甲基亚硝胺标准工作溶液。
2 结果与分析
2.1 条件优化
2.1.1 提取液的选择
提取溶剂对检测结果有较大影响。在空白样品中加入100 ng N-二甲基亚硝胺,分别考察乙腈、二氯甲烷、正己烷3种提取溶剂对空白样品提取效果的影响。试验重复3次。结果表明,乙腈的提取效果最佳;其次是二氯甲烷;正己烷的提取效果较差,难以满足定量要求。
2.1.2 氮吹水浴温度的选择
N-二甲基亚硝胺的相对分子质量较小、性质不稳定,氮吹水浴温度直接影响目标化合物的回收率。在加标量为20 ?g·kg-1的条件下,采用加标回收试验的方式,对10 ℃、15 ℃、20 ℃、30 ℃4个温度条件进行考察。由表2可知,当氮吹水浴温度为15 ℃时,氮吹的时间较短,回收率最大。当氮吹水浴温度小于15 ℃时,回收率虽然较大,但氮吹时间过长;当氮吹水浴温度大于15 ℃时,随着水浴温度的升高,目标化合物的挥发较快,导致回收率下降。
2.1.3 仪器条件的优化
(1)洗脱条件的优化。为实现目标物与其他干扰物质的良好分离,对仪器流动相洗脱条件进行了优化。采用某空白加标样品作为测试样,分别考察不同洗脱条件(0.1%甲酸水溶液 - 甲醇、0.1%甲酸水溶液-0.1% 甲酸甲醇、0.2%甲酸水溶液-甲醇、0.1%甲酸水溶液-乙腈)下的洗脱效果。结果表明,0.1%甲酸水溶液-0.1%甲酸甲醇为流动相时,分离度良好,洗脱效率高。
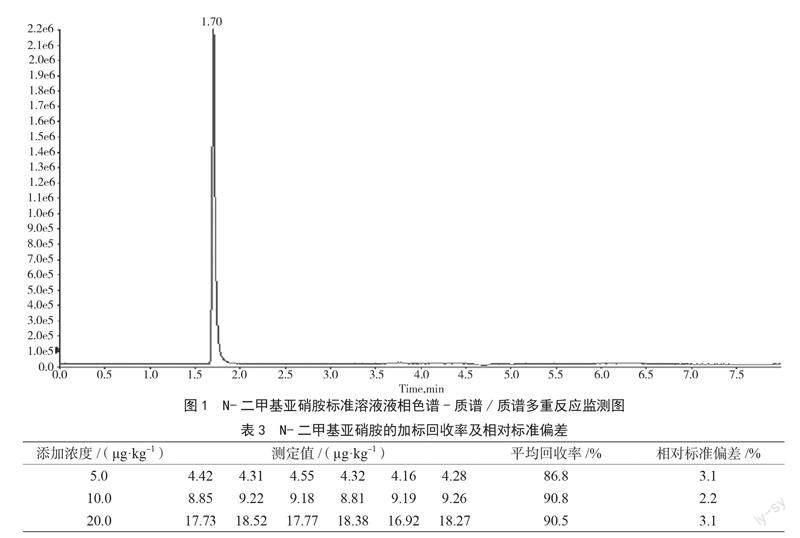
(2)色谱柱的优化。对Waters XBridge C18、Waters Atlantis T3、Waters UPLC BEH C18 3种色谱柱的分离效果进行比较。结果显示,当采用Waters XBridge C18柱时,目标化合物分离好,峰形尖锐,保留时间适中,能满足定量检测要求。因此,最终选择Waters XBridge C18色谱柱。N-二甲基亚硝胺标准溶液液相色谱-质谱/质谱多重反应监测图如图1所示。
2.2 方法学考察
以目标化合物峰面积比为纵坐标,质量浓度为横坐标绘制标准曲线。结果显示,标准曲线线性范围为2.0~200.0 ng·mL-1,线性回归方程为y=7 575.77x+6 345.784 3,相关系数为0.998 5,表明标准曲线线性关系良好。信噪比为3时,检出限为0.5 μg·kg-1;信噪比为10时,定量限为3.0 μg·kg-1。
2.3 加标回收试验
选取熏煮火腿作为样品,添加N-二甲基亚硝胺标准品,设置5.0 μg·kg-1、10.0 μg·kg-1、20.0 μg·kg-1 3个加标水平,每个水平6个平行,按1.2处理后进行测定,分别计算加标回收率与相对标准偏差。由表3可知,不同加标浓度下,N-二甲基亚硝胺的平均回收率为86.8%~90.8%,相对标准偏差为2.2%~3.1%。结果表明,该方法准确高,稳定性好。
2.4 实际样品分析
采用本方法对本地区2种不同来源的肉制品(香肠、腌鱼),共16个样品进行分析。部分样品中检出N-二甲基亚硝胺留,含量为0~16.4 μg·kg-1,表明样品中有一定量的N-二甲基亚硝胺残留。
3 结论
本研究建立了一种QuEChERS结合液相色谱-质谱联用法筛查、定性确证与定量动物源食品中N- 二甲基亚硝胺的方法。本方法在2.0~200.0 ng·mL-1具有良好的线性关系,检出限为0.5 μg·kg-1,定量限为3.0 μg·kg-1,回收率在86.8%~90.8%,优化了前处理条件,省去了蒸馏、萃取、旋蒸等复杂的前处理步骤,简化了提取和净化过程,缩短了实验时间,方法简单高效,为N-二甲基亚硝胺测定提供了一种新的可靠的方法。
参考文献
[1]谢庆超,王子,李銀辉,等.发酵肉制品中的危害因素及防控措施研究进展[J].食品科学,2023,44(19):230-238.
[2]田建军,张开屏,景智波,等.发酵肉制品加工中衍生的非健康因子控制研究进展[J].中国食品学报,2020,20(1):275-283.
[3]GUSHGARI J A,HALDEN U R.Critical review of major sources of human exposure to N-nitrosamines.[J].Chemosphere,2018,210:1124-1136.
[4]HERRMANN S,GRANBY K,DUEDAHL-OLESEN L.Formation and mitigation of N-nitrosamines in nitrite preserved cooked sausages[J].Food Chemistry,2015,174:516-526.
[5]李雅,郝桂娟,黄程,等.2018~2020年国家食品安全监督抽检不合格数据分析[J].食品与药品,2022,24(3):
256-261.
[6]王娟强,齐婧,李贺楠,等.发酵肉制品食品安全风险分析及监管建议[J].肉类研究,2021,35(8):54-63.
[7]黄仁贵,马龙,谭莎莎,等.肉制品中N-二甲基亚硝胺检测方法的研究进展[J].粮食与油脂,2023,36(6):1-4.
[8]陈婧,王立媛,胡争艳,等.即食水产制品N-亚硝胺类化合物检测的样品处理方法优化研究[J].预防医学,2023,35(8):726-731.
[9]LONA-RAMIREZ J F,GONZALEZ-ALATORRE G,RICO-RAM?REZ V,et al.Gas chromatography/mass spectrometry for the determination of nitrosamines in red wine[J].Food chemistry,2016,196:1131-1136.
[10]陶声萍.QuEChERS-GC-MS/MS法快速检测食品中N-二甲基亚硝胺[J].现代农业科技,2023(7):198-201.
作者简介:赵广西(1979—),男,河北秦皇岛人,硕士,副高级工程师。研究方向:食品安全检测。
