火锅底料中喹诺酮类化合物的测定
2024-04-15康宗华江露
康宗华 江露

摘 要:目的:建立火锅底料中喹诺酮类化合物的测定方法。方法:样品经0.1 mol·L-1 EDTA-Mcllvaine溶液提取,5%甲醇水溶液净化后上液相色谱-串联质谱仪分析。采用SHIMADZU C18色谱柱(2.1 mm×100 mm,3 ?m)进行分离,流动相为0.1%甲酸-乙腈,梯度洗脱。采用电喷雾离子源,正离子扫描。结果:该方法线性关系良好,相关系数≥0.99;样品在1.0~20.0 μg·kg-1的添加水平下加标回收率为65.20%~110.20%。结论:该方法适用于火锅底料中喹诺酮类化合物的测定,简单快速,灵敏度高,重复性好。
关键词:液相色谱-串联质谱法;火锅底料;喹诺酮
Determination of Quinolones in Hot Pot Base
KANG Zonghua1,2, JIANG Lu1
(1.Hunan Kouweiwang Group Co., Ltd., Yiyang 413000, China;
2.Hunan Tuopu Inspection Institute, Yiyang 413000, China)
Abstract: Objective: To establish a method for the determination of quinolones in hot pot base. Method: Hot pot base sample were extracted by 0.1 mol·L-1 EDTA-Mcllvaine solution, purified by 5% methanol aqueous solution, and analyzed by liquid chromatography-tandem mass spectrometry. Separation was performed using a column SHIMADZU C18 column (2.1 mm×100 mm, 3 ?m) with 0.1% formic acid-acetonitrile as a mobile phase and gradient elution. The ion source is an electrospray ion source, and the scanning method is positive ion scanning. Result: The method has a good linear relationship, and the correlation coefficient is greater than or equal to 0.99. The recoveries of spiked samples at spiked levels of 1.0~20.0 μg·kg-1 ranged from 65.20% to 110.20%. Conclusion: This method is suitable for the determination of quinolones in hotpot seasonings, which is simple, rapid, sensitive and reproducible.
Keywords: liquid chromatography-tandem mass spectrometry; hot pot base; quinolones
近年来,多地质监局在一些商贩所用的火锅底料中检出了喹诺酮类化合物[1]。由于喹诺酮类化合物对革兰氏阴性菌具有杀菌作用,被广泛用作人畜通用的抗生素药物,可用于治疗革兰氏阴性菌感染的大部分疾病。但根据相关规定,洛美沙星等4种兽药在食品动物中已经被禁止使用[2]。喹诺酮类化合物会对胃肠道、中枢神经产生不良作用。例如,氧氟沙星是一种喹诺酮类化合物,长期食用氧氟沙星残留量超标的动物性食品可能会引发胃肠道不良反应,包括刺激性损伤和一些不适症状(如头痛、睡眠不良)等,严重者还可能引起肝损害[3]。目前,喹诺酮类化合物的检测方法主要有液相色谱[4]、液质联用[1,5]等,但这些检测方法参数不确定,重复性不佳。因此,本研究旨在采用液相色谱-串联质谱法,为喹诺酮类化合物的高效檢测提供方法依据。
1 材料与方法
1.1 材料与试剂
火锅底料,从超市购买;甲醇,优级纯,默克;乙腈,优级纯,汇虹;甲酸、氨水,分析纯,汇虹;磷酸氢二钠、盐酸、乙酸铵,分析纯,国药集团;NaOH、EDTA-2Na,分析纯,科密欧。除有特殊说明,本文所用试剂均为分析纯,试验用水均为超纯水。
1.2 仪器与设备
AB3500液质联用仪,Sciex TRIPLE QUAD TM3 500;Ax224ZH电子天平,奥豪斯;SHZMADZU C18色谱柱(2.1 mm×100 mm,3 μm)。
1.3 试剂与标准溶液配制
0.1 mol·L-1 EDTA Mcllvaine缓冲溶液(以下简称缓冲溶液):称取60.5 g EDTA-2Na至1 625 mL Mcllvaine缓冲溶液中,超声溶解。25%氨水甲醇溶液:量取25 mL氨水于100 mL容量瓶中,用甲醇定容至刻度线。
标准储备液(有效期6个月):称取各标准品约10 mg(精确到0.001 g)于10 mL容量瓶中,加入0.2 mL甲酸,超声溶解,乙腈定容,得到浓度为1.0 mg·mL-1的标准储备液,避光保存(<18 ℃)。
混合标准溶液(有效期3个月):量取一定量各标准储备液,用甲醇稀释,使环丙沙星、沙拉沙星浓度为0.015 mg·mL-1,其他化合物浓度为0.010 mg·mL-1。
混合标准中间溶液:量取混合标准溶液1 mL于10 mL容量瓶中,用甲醇精确稀释至刻度线,得到环丙沙星、沙拉沙星浓度为1.5 μg·mL-1,其他化合物浓度为1.0 μg·mL-1的混合标准中间溶液,避光保存(<4 ℃),现用现配。
内标标准储备液(有效期6个月):称取氘代同位素标准品10 mg,加入0.2 mL甲酸,超声溶解,用乙腈定容至10 mL,得到浓度为1.0 mg·mL-1的内标标准储备液,避光保存(<18 ℃)。
1.4 实验方法
1.4.1 样品处理
火锅底料样品制备:称取待检样品约500 g,用绞肉机充分绞碎均匀,装入干净容器中,密封,做好相关信息标记,4 ℃冷藏存放。
1.4.2 样品制备
样品制备参考相关标准方法[6]。
(1)提取。称取5.0 g均匀样品(精确至0.001 g)于50 mL离心管中,加入混合内标标准中间溶液0.1 mL,加入20 mL 0.1 mol·L-1 EDTA-Mcllvaine缓冲溶液,涡旋混匀1 min,超声提取15 min,8 000 r·min-1离心5 min,将上清液转移至50 mL容量瓶中,残渣用EDTA-Mcllvaine缓冲溶液重复提取两次,每次10 mL,合并上清液至同一容量瓶中,用EDTA-Mcllvaine缓冲溶液定容至刻度,混匀。用滤纸过滤(必要时10 000 r·min-1离心5 min后过滤),续滤液待净化。
(2)净化。精密吸取上述待净化液25.0 mL,以1 mL·min-1的流速全部通过固相小柱,用3 mL 5%甲醇水溶液淋洗,抽干,依次以6 mL甲醇、3 mL氨水甲醇洗脱,合并甲醇及氨水甲醇洗脱液,45 ℃氮吹至近干,准确加入2.0 mL流动相初始比例复溶液溶解残渣,过0.22 ?m滤膜,滤液供液相色谱-串联质谱仪分析。
1.4.3 标准曲线配制
基质标准工作溶液的制备:称取5 g空白试样(精确至0.001 g)于50 mL离心管中,除不加入混合内标标准中间溶液外,其余操作同1.4.2中样品处理,得到空白基质,共制备6份。
分别准确吸取混合标准中间溶液0 mL、0.010 mL、0.020 mL、0.050 mL、0.100 mL、0.200 mL及混合内标标准中间溶液0.025 mL于试管中,氮吹至近干,加入1.0 mL空白基质复溶,过0.22 μm滤膜,得到浓度分别为0 ng·mL-1、10 ng·mL-1、20 ng·mL-1、50 ng·mL-1、100 ng·mL-1、200 ng·mL-1的基质标准工作溶液用以上机。用内标法绘制标准曲线。
1.4.4 测试条件
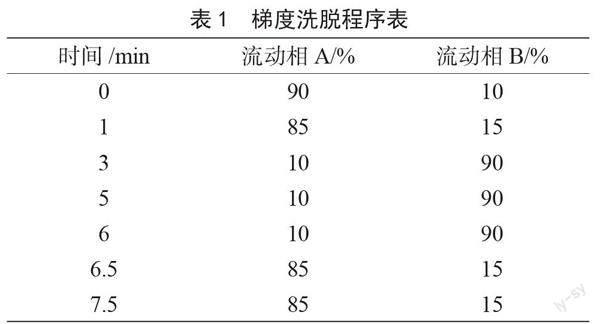
色谱柱:SHIMADZU C18柱(2.1 mm×100 mm,3 ?m);流动相:A-0.1%的甲酸水溶液,B-乙腈;经过前期多次预实验,最终优化得到梯度洗脱程序见表1;流速:300 μL·min-1;柱温:40 ℃;进样量:20 μL。
质谱条件:电喷雾离子源;采用正离子扫描;进行多反应监测;电喷雾电压:-4 500 V;喷雾气(GS1)压力:30 psi;气帘气压力:10 psi;辅助加热气(GS2):28 psi;离子源温度:350℃;碰撞气:8 psi。
1.4.5 加标回收试验和精密度试验
(1)加标回收试验。制备高、中、低3个加标水平的火锅底料样品,按照样品前处理方法提取、净化后,按1.4.4条件测定加标回收率。
(2)精密度试验。进行两次平行实验,计算测定结果的绝对差值。
2 结果与分析
2.1 线性相关性和方法检出限
分别称取样品8份,按照1.4处理。由表2可知,11种喹诺酮类化合物在0~200 ng·mL-1线性关系良好。检出限为1 ?g·kg-1,定量限为3 ?g·kg-1。
2.2 回收率和重复性
由表3可知,样品在1.0~20.0 μg·kg-1添加水平下的加标回收率为65.20%~110.20%。
2.3 样品检测
对从超市购买的火锅底料进行喹诺酮类化合物含量测定,结果显示未检出。
3 结论
本实验建立了高效液相色譜-串联质谱法测定火锅底料中喹诺酮类化合物的方法,应用此方法对实际样品中喹诺酮类化合物进行测定,结果为未检出。该方法简单快速,灵敏度高,重复性好,可推广应用于更多食品中喹诺酮类化合物的检测,如麻辣烫、豆制品等。
参考文献
[1]赵飞,高广慧,贾宏新,等.超高效液相色谱-串联四极杆质谱法测定麻辣烫中喹诺酮类药物残留[J].分析测试学报,2017,36(6):768-772.
[2]佚名.市场监管总局关于发布《水产品及水中丁香酚类化合物的测定》等2项食品补充检验方法的公告(2019年第15号)[J].中国食品卫生杂志,2019,31(3):204.
[3]史艳艳,高琳,李俊,等.液相色谱-串联质谱法检测饲料中洛美沙星、培氟沙星、氧氟沙星、诺氟沙星残留[J].食品安全质量检测学报,2019,10(11):3367-3375.
[4]重庆市食品药品检验检测研究院.香辛料及其制品中真菌毒素的检测方法:202111678178.8[P].2022-04-05.
[5]温颖婉,黎艳,孙树周.用液相色谱-串联质谱法同时检测化妆品中17种喹诺酮类抗生素方法的研究[J].当代医药论丛,2019,17(9):220-222.
[6]国家市场监督管理总局.豆芽、豆制品、火锅及麻辣烫底料中喹诺酮类、磺胺类、硝基咪唑类、四环素类化合物的测定:BJS 202310[S].北京:中国标准出版社,2023.
