一例遗传性骨髓衰竭综合征中RTEL1基因新突变的研究
2023-09-07杨立雪蔡海燕闫金松卢莹
杨立雪,蔡海燕,闫金松,卢莹
(1.上海交通大学医学院附属新华医院,上海交通大学医学院皮肤病研究所,上海 200092; 2.上海交通大学医学院细胞分化与凋亡教育部重点实验室病理生理学教研室,上海 200092; 3.辽宁省造血干细胞移植医学中心血液科,辽宁省造血干细胞移植与转化医学重点实验室,大连血液学重点实验室,大连医科大学第二附属医院钻石湾血液学研究所,辽宁 大连 116000)
0 引言
遗传性骨髓衰竭综合征是一组血液系统的遗传性疾病,其共同的临床特征为骨髓造血功能衰竭,患者先天发育异常,并且具有发展为恶性疾病的风险[1-2].遗传性骨髓衰竭综合征包括先天性角化不良、范可尼贫血、舒-戴二氏综合征、戴-布二氏贫血等[1,3].由于遗传性骨髓衰竭综合征的发病率较低,并且临床表现呈多样性,因此传统的检测和治疗手段有限.随着下一代测序的发展,越来越多的临床医生将基因检测技术运用到临床诊断与治疗当中,使人们对于该疾病的认识更加深入,对病人的及时诊断、有效治疗及预后改善等方面在临床都得到了更大的提高.
其中,先天性角化不良(dyskeratosis congenia, DC)是一种先天性遗传性疾病,是一种具有遗传异质性的骨髓衰竭综合征[1].经典的DC临床特点表现为三联征:甲营养不良、皮肤异常色素沉着,以及口腔黏膜白斑[4].其中,骨髓衰竭是目前为止DC患者最常见的死亡原因[5].DC病情的严重程度在个体之间差异较大,表型较轻者进表现为三联征,表型严重者在儿童期便会死亡[6].该病具有遗传异质性,主要有伴X染色体隐性、常染色体显性和常染色体隐性三种遗传方式[4].由于DC属于罕见病,DC患者的临床表现具有多样性,并且具有遗传异质性,因此DC的临床确诊具有一定的难度,对DC的及时准确确诊至关重要.目前的研究表明,引起先天性角化不良的主要原因是端粒缩短和复制老化,导致细胞早衰以及组织损伤[4, 7].
RTEL1(regulator of telomere length 1)是superfamily 2 DEAH子家族中含Fe-S簇的一种重要的DNA解旋酶[8].RTEL1作为DNA解旋酶,在DNA复制时通过解开端粒上的t环和G4四链体(DNA二级结构),促进DNA复制、修复和重组,维持基因组的完整性,并起到保护端粒的作用[9-12].据报道,RTEL1的双等位基因突变,无论是纯合或杂合突变,都会引起端粒长度缩短,临床表现为儿童早期的先天性角化不良和Hoyeraal-Hreidarsson (HH) 综合征[13-17].
本研究基于临床上一例男性儿童遗传性骨髓衰竭综合征DC患者展开.通过收集该患者外周血样本,并针对于与先天性骨髓衰竭密切相关的69个基因的全部外显子区域和旁侧内含子区域进行测序,发现该患者的RTEL1基因编码序列的第540个氨基酸由缬氨酸突变为异亮氨酸(V540I).本研究构建了表达稳定该突变的细胞系,在体外对其功能和分子机制进行了初步探索.
1 实验材料与方法
1.1 实验材料
1.1.1 细胞 本研究使用的293细胞(人胚肾细胞)购自中国科学院典型培养物保藏委员会细胞库.经鉴定,该细胞系无支原体污染.
1.1.2 主要试剂 胎牛血清、高糖DMEM培养基购于美国Gibco公司,青霉素-链霉素双抗购于苏州新赛美生物科技有限公司,Lipofectamine 2000转染试剂、新霉素购于美国Thermo Fisher Scientific公司,psPAX2质粒、pMD2G质粒购于美国Addgene公司,polybrene购于美国Sigma-Aldrich公司,细胞计数试剂盒8(cell counting kit-8,CCK-8)购于日本DOJINDO公司,快速银染试剂盒、细胞衰老β-半乳糖苷酶染色试剂盒购于上海碧云天生物技术有限公司,STA-9090购于美国MCE公司,Anti-FLAG M2 beads购于美国Thermo Fisher Scientific公司.
实验中所用到的抗体,anti-flag抗体购于美国Thermo Fisher Scientific公司,anti-RTEL1抗体购于武汉三鹰生物技术有限公司,辣根过氧化物酶(HRP)标记羊抗小鼠二抗、HRP标记羊抗兔二抗购于德国Merck millipore公司,Alexa Fluro 488标记的山羊抗鼠二抗购于美国Invitrogen公司,β-actin抗体购于日本MBL公司.
1.2 实验方法
1.2.1 293细胞培养 293细胞为贴壁细胞,培养于含有10%胎牛血清和1%青霉素-链霉素双抗的高糖DMEM培养基中,于37 ℃、含5% CO2的培养箱中恒温培养,每2~3 d传代一次.
1.2.2 过表达RTEL1-WT和RTEL1-MUT的293细胞系的构建 通过转染试剂Lipofectamine 2000将慢病毒包装体系psPAX2质粒、pMD2G质粒、以及pLVneo-RTEL1-WT质粒、pLVneo-RTEL1-MUT质粒转染至293T细胞,分别于转染后48 h和72 h收取细胞上清病毒液,添加polybrene感染293细胞.于感染后72 h添加2 000 μg/mL的新霉素,筛选出稳定表达RTEL1-WT和RTEL1-MUT的293细胞.
1.2.3 免疫荧光实验 将细胞种于放置了鹏硅酸盐处理的玻片的皿中,待细胞密度约为60%~70%时,用PBS洗涤两遍.加入4%多聚甲醛室温固定细胞15 min,PBS洗3次,每次5 min.加入0.2% TritonX-100室温破膜15 min,PBS洗3次,每次5 min.加入3% BSA室温封闭30 min后,一抗4 ℃孵育过夜.次日用PBS洗3次,每次5 min.二抗室温避光孵育1 h,PBS洗3次,每次5 min.最后加入DAPI进行封片,于Nikon A1R激光共聚焦显微镜下观察并拍照.
1.2.4β-半乳糖苷酶细胞衰老实验β-半乳糖苷酶细胞衰老实验步骤按照上海碧云天生物技术有限公司细胞衰老β-半乳糖苷酶染色试剂盒说明书进行.简言之,用PBS洗涤六孔板中的细胞后,加入β-半乳糖苷酶染色固定液室温固定15 min,PBS洗3次,每次3 min.加入染色工作液,于二氧化碳培养箱中37 ℃孵育过夜,于普通光学显微镜下观察并拍照.
1.2.5 细胞增殖实验 于96孔板中每孔种1.5×103个细胞,并设置仅加培养基的空白对照组,每组设置3副孔.24 h待细胞贴壁后,每孔加入10 μL CCK-8试剂,于37 ℃、含5% CO2的培养箱内孵育2 h后,用酶标仪测定450 nm处的吸光度,将减去空白对照组后的数值作为Day 0的吸光度值.之后每24 h检测一次吸光度值,除以Day 0的吸光度值以计算细胞数的变化倍数,绘制生长曲线.
1.2.6 Western Blot实验 将等量蛋白样品上样至 10% SDS-聚丙烯酰胺凝胶孔中,然后转膜至硝酸纤维素膜上.用 2% BSA室温封闭1 h,将膜与一抗4 ℃孵育过夜,第二日用PBS洗膜3次,每次5 min.用HRP连接的二抗室温孵育1 h.通过化学发光 phototope-HRP 试剂盒进行检测.
1.2.7 免疫沉淀实验 收集5×107个细胞,PBS洗两遍之后,加入NP40裂解液,充分裂解后15 000 g离心10 min,取上清.取1/10上清作为Input样本,剩余裂解液加入50 μL M2 beads,于4 ℃孵育过夜.第二日用NP40洗beads 3次,PBS洗beads 1次,每次5 min. 500 g离心1 min,弃上清,加入100 μL 2×SDS裂解液,获得免疫沉淀样品.
1.2.8 银染实验 银染实验按照上海碧云天生物技术有限公司快速银染试剂盒说明书操作步骤进行.简言之,将蛋白样品上样至 10% SDS-聚丙烯酰胺凝胶上,电泳结束后加入固定液室温固定1 h.用30%乙醇和双蒸水洗涤后,加入增敏液室温孵育2 min.用双蒸水洗涤2次后,加入银染液室温孵育10 min.用双蒸水洗涤后,加入显色液,待出现比较理想的预期蛋白条带时,加入银染终止液.最后用双蒸水洗涤后,可在双蒸水中保存.
1.2.9 统计学分析 使用Graphpad Prizm 8.0.1软件进行绘图分析以及数据处理.通过标准双尾Student’st检验比较两组独立样本.所有实验至少重复3次.*,P<0.05;**,P<0.01;***,P<0.001.认为P<0.05具有统计学意义.
2 实验结果与分析
2.1 在遗传性骨髓衰竭综合征患者中发现RTEL1基因突变本研究基于临床上的一例男性儿童遗传性骨髓衰竭综合征患者展开.该患者血常规显示:白细胞1.65×109/L、中性粒细胞绝对值0.07×109/L、血红蛋白59 g/L.骨髓细胞学提示:骨髓增生低下,粒系增生受抑,红系增生不良,成熟红细胞轻度大小不一,淋巴细胞比例增高,少数胞浆可见颗粒、有毛刺突起,全片未见巨核细胞,诊断为再生障碍性贫血骨髓象.患者周身皮肤见和色斑片及大片色素沉着,部分可见细网状,考虑与先天性角化不良有关.胸部CT影像学表现:双肺纹理增粗,透过度略减低,左肺为著.影像诊断:双肺轻度间质性转变,左肺为著.通过对该患者外周血样本针对于先天性骨髓衰竭密切相关的69个基因的全部外显子区域和旁侧内含子区域进行测序,发现该患者的RTEL1基因编码序列的第540个氨基酸由缬氨酸突变为异亮氨酸(V540I)(图1).经家系验证,发现患者父亲该位点为杂合,母亲该位点为野生型纯合(图1).患者最终诊断为遗传性骨髓衰竭综合征,具体表现为先天性角化不良.

图1 男性儿童遗传性骨髓衰竭综合征患者遗传学表现遗传性骨髓衰竭综合征患者及其父母的外周血全部外显子区域和旁侧内含子区域测序结果.左图:母亲;中间图:父亲;右图:患者.由测序结果可以看出,该患者的RTEL1基因编码序列的第540个氨基酸由缬氨酸突变为异亮氨酸(V540I).
2.2 建立过表达RTEL1-WT和RTEL1-MUT的293细胞系RTEL1是一种重要的DNA解旋酶.RTEL1蛋白上有多个功能结构域,包括位于7~320氨基酸序列的解旋酶ATP结合的Helicase ATP-binding结构域,位于111~296氨基酸序列的ATP结合和水解所必须的DEAD_2结构域,位于471~767氨基酸序列的具有5‘至3’方向性的DNA解旋酶DNA repair helicase结构域,位于901~1 000氨基酸序列的可能充当蛋白与蛋白相互作用的分子支架的harmonin-N-like结构域,以及已在体内验证用于RTEL1-PCNA相互作用的PIP结构域[9](图2A).此外,对RTEL1蛋白结构的分析结果表明,V540其周围距离较近的氨基酸大多数为疏水氨基酸,有VAL765、 PHE733、PHE762.因此,V540与这些氨基酸可能形成一个疏水的相互作用(图2B左图).当V突变成I后,I与VAL765的原子之间距离过近,有可能不利于蛋白整体的稳定,可能会导致局部甚至整体的构象不稳定(图2B右图).患者RTEL1的V540I突变位于DNA repair helicase结构域,因此推测该突变可能通过影响RTEL1的DNA解旋酶功能,从而导致先天性角化不良的发生.为了进一步探索RTEL1的V540I突变对RTEL1功能的影响,首先我们在带有新霉素抗性的质粒pLVneo中插入带有flag标签的RTEL1-WT和RTEL1-MUT序列,将质粒转染至人胚肾细胞系293细胞中,并用新霉素筛选出稳定表达RTEL1-WT-flag和RTEL1-MUT-flag的293细胞.通过Western Blot检测flag的表达,结果显示,在293细胞中检测到了flag条带,证明成功构建了过表达RTEL1-WT和RTEL1-MUT的293细胞系(图2B).
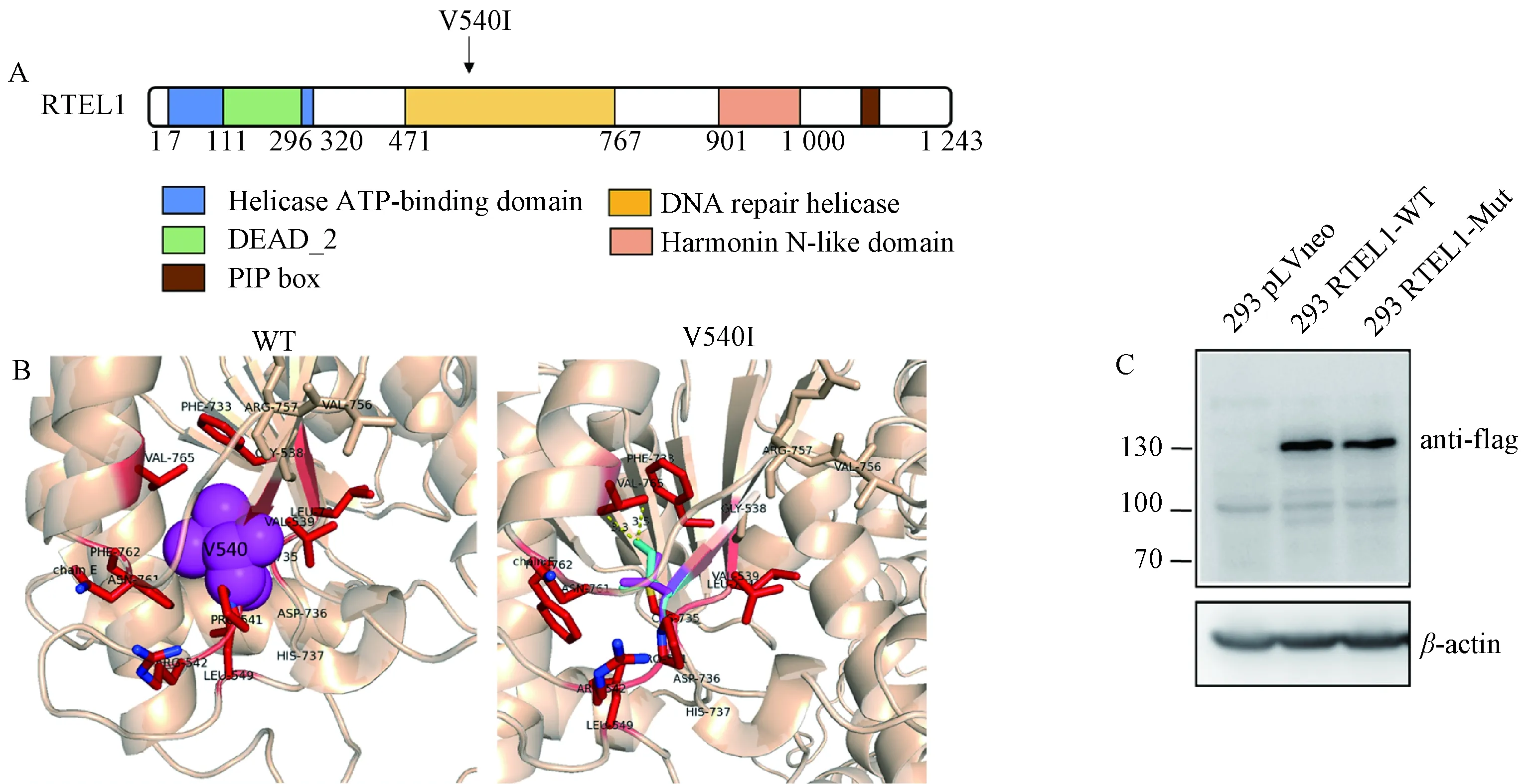
图2 过表达RTEL1-WT和RTEL1-MUT的293细胞系的构建A RTEL1蛋白结构域示意图.如图所示,完整的RTEL1包括位于7~320氨基酸序列的解旋酶ATP结合的Helicase ATP-binding结构域,位于111~296氨基酸序列的ATP结合和水解所必须的DEAD_2结构域,位于位于471~767氨基酸序列的具有5‘至3’方向性的DNA解旋酶DNA repair helicase结构域,位于901~1000氨基酸序列的可能充当蛋白与蛋白相互作用的分子支架的harmonin-N-like结构域,以及已在体内验证用于RTEL1-PCNA相互作用的PIP结构域.B RTEL1-WT和RTEL1-MUT的540号氨基酸附近的蛋白结构.左图:V540其周围距离较近的氨基酸大多数为疏水氨基酸,有VAL765、PHE733、PHE762.因此,V540与这些氨基酸可能形成了一个疏水的相互作用.右图:当V突变成I后,I与VAL765的原子之间距离过近,有可能不利于蛋白整体的稳定,可能会导致局部甚至整体的构象不稳定.C Western Blot结果显示在过表达RTEL1-WT和RTEL1-MUT的293细胞系中检测到了flag条带,证明成功构建了过表达RTEL1-WT和RTEL1-MUT的293细胞系.
2.3 RTEL1-MUT的表达抑制293细胞的增殖为了探究过表达RTEL1-WT和RTEL1-MUT对293细胞的影响,我们展开了一系列的功能实验.首先,我们想要探究RTEL1-WT和RTEL1-MUT的定位是否存在差异.我们使用anti-flag抗体,通过免疫荧光技术标记293细胞中外源性的RTEL1-WT和RTEL1-MUT.免疫荧光结果显示,外源性的RTEL1-WT和RTEL1-MUT基本定位于胞浆中,二者的定位并无明显差别(图3A).第二,考虑到已有文献报道RTEL1于PIP结构域的突变会引起细胞衰老[18],因此我们通过β-半乳糖苷酶衰老染色检测了过表达RTEL1-WT和RTEL1-MUT的293细胞系的衰老情况,结果显示两种细胞系均无明显衰老情况(图3B).由于RTEL1作为与维持端粒相关的DNA解旋酶,其活性对细胞的复制潜能非常重要[19],且V540I位于RTEL1的DNA解旋酶结构域,因此我们推测RTEL1-MUT的表达可能会影响细胞的增殖能力.我们将过表达RTEL1-WT和RTEL1-MUT的293细胞种于96孔板中,使用CCK-8试剂盒,每隔24 h测量一次OD450,连续测量4 d.以第0 d的OD450为基准,计算每24 h细胞数的变化倍数.结果显示,在相同的培养时间下,过表达RTEL1-MUT的细胞增殖速率明显低于过表达RTEL1-WT的293细胞,表明RTEL1-MUT的表达会抑制293细胞的增殖(图3C).
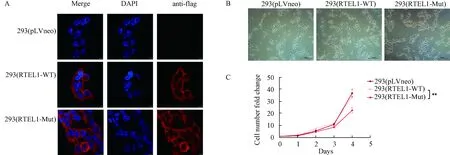
图3 过表达RTEL1-WT和RTEL1-MUT的293细胞系功能实验A 免疫荧光检测过表达RTEL1-WT和RTEL1-MUT的293细胞系中外源性RTEL1的定位. 外源性的RTEL1-WT和RTEL1-MUT基本定位于胞浆中,二者的定位并无明显差别. B β-半乳糖苷酶衰老染色检测过表达RTEL1-WT和RTEL1-MUT的293细胞系的衰老情况,结果显示两种细胞系均无明显衰老情况. C CCK-8试剂盒检测RTEL1-WT和RTEL1-MUT的293细胞系的增殖速率,过表达RTEL1-MUT的细胞增殖速率明显低于过表达RTEL1-WT的293细胞.通过标准双尾Student’s t检验比较两组独立样本.**,P<0.01.认为P<0.05具有统计学意义.
2.4 RTEL1-MUT与HSP90的相互作用减少为进一步了解RTEL1-MUT抑制293细胞增殖的机制,我们希望探究与RTEL1-WT和RTEL1-MUT相互作用存在差异的蛋白.首先,我们采用免疫沉淀技术(immunoprecipitation, IP),使用anti-flag抗体,对293细胞中的RTEL1-WT和RTEL1-MUT蛋白进行富集.接着,我们通过Western Blot技术对富集后的蛋白进行了验证(图4A).在IP样品中,用anti-flag和anti-RTEL1抗体都可检测到条带,证明RTEL1-WT和RTEL1-MUT蛋白被成功地特异性富集.随后,我们采用银染技术,以确定与RTEL1-WT和RTEL1-MUT相互作用存在差异的蛋白,发现在100 kD附近存在一个条带,该条带在RTEL1-WT泳道中比在RTEL1-MUT泳道中深(图4B).我们通过质谱技术对该条带的蛋白进行探究,根据质谱结果将目光锁定在HSP90上.目前已有报道表明,HSP90作为伴侣蛋白可以与端粒酶复合物结合并起到维持其活性的作用[20],但HSP90与RTEL1的相互作用目前尚少有人报道.
接下来,我们通过Western Blot技术对质谱结果进行了验证.在IP样品中,用anti-HSP90抗体可以检测到条带,证实HSP90与RTEL1存在相互作用,并且与RTEL1-WT相互作用的HSP90明显多于与RTEL1-MUT相互作用的HSP90,说明RTEL1的V540I突变会影响RTEL1与HSP90的相互作用,并使得RTEL1-MUT与HSP90的相互作用减少(图4C).
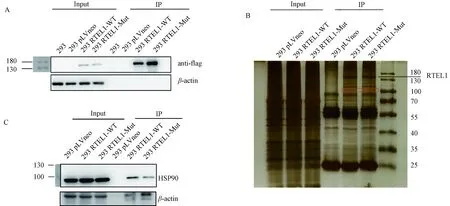
图4 RTEL1突变影响其与HSP90的相互作用A 使用anti-flag抗体,对293细胞中的RTEL1-WT和RTEL1-MUT蛋白进行免疫沉淀富集,通过Western blot检测免疫沉淀样品,结果显示在免疫沉淀样品中显著富集到了带flag标签的野生型和突变型RTEL1. B 免疫沉淀样品银染结果.箭头指示富集到的RTEL1蛋白.红色框圈出部分为100 kD附近野生型和突变型RTEL1免疫沉淀样本中存在差异的条带.C 免疫沉淀样品Western Blot结果.在免疫沉淀样品中检测到了HSP90,证实了HSP90与RTEL1的相互作用,并且与RTEL1-WT相互作用的HSP90明显多于与RTEL1-MUT相互作用的HSP90.
2.5 HSP90与RTEL1相互作用以维持其稳定性为了深入了解HSP90对RTEL1功能的影响,我们使用200 nmol/L HSP90抑制剂STA-9090分别处理过表达RTEL1-WT和RTEL1-MUT的293细胞,并于处理后0、6、12、24 h收取细胞裂解液蛋白.Western Blot结果显示,在用STA-9090处理后6 h,RTEL1蛋白水平便有明显下降(图5).STA-9090可以结合于HSP90的N末端ATP结合域,从而诱导HSP90受体蛋白的降解[21-22].该结果进一步证实了HSP90与RTEL1的相互作用,并且HSP90可以维持RTEL1蛋白的稳定性.
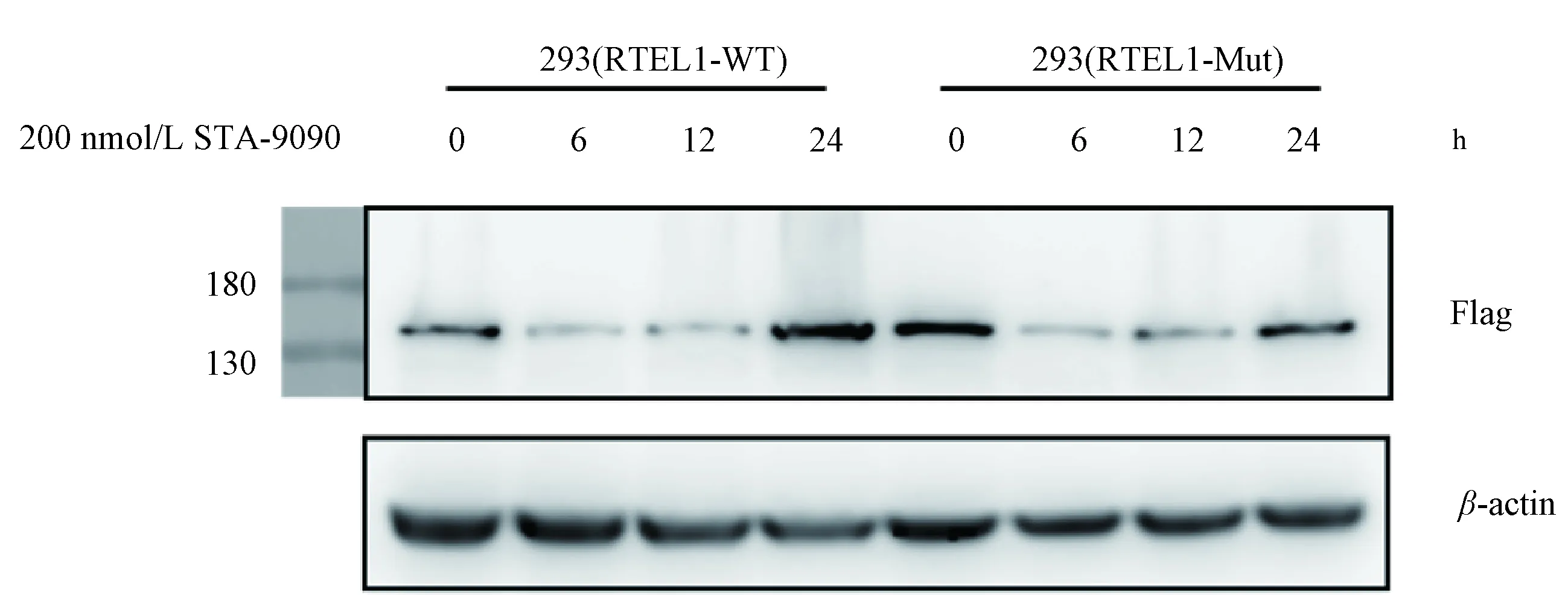
图5 200 nmol/L HSP90抑制剂STA-9090时间梯度处理过表达RTEL1-WT和RTEL1-MUT的293细胞蛋白western blot结果200 nmol/L STA-9090处理6 h后,RTEL1蛋白水平明显下降.
3 讨论
由于遗传性骨髓衰竭综合征较为罕见且临床表现多样性,传统的治疗检测手段有限.其中,先天性角化不良是骨髓衰竭综合征的一种,具有遗传异质性[1].由于DC属于罕见病,DC患者的临床表现具有多样性,并且具有遗传异质性,因此DC的临床确诊具有一定的难度,对DC的及时准确确诊至关重要.
本研究旨在探究DNA解旋酶RTEL1突变导致先天性角化不良发生的机制,以及RTEL1的生物学功能,将下一代测序技术运用到对遗传性骨髓衰竭综合征的临床诊断上.本研究基于临床上一例男性儿童先天性角化不良患者展开,通过收集该患者外周血样本并针对于先天性骨髓衰竭密切相关的69个基因的全部外显子区域和旁侧内含子区域对该样本进行测序,发现该患者的RTEL1基因编码序列的第540个氨基酸由缬氨酸突变为异亮氨酸(V540I)外显子. 患者RTEL1的V540I突变位于DNA repair helicase结构域,因此推测该突变可能通过影响RTEL1的DNA解旋酶功能,从而导致先天性角化不良的发生.为了进一步探索RTEL1的V540I突变对RTEL1功能的影响,我们成功构建了过表达野生型RTEL1(RTEL1-WT)以及突变型RTEL1(RTEL1-MUT)的293细胞系.接下来,我们在所构建的过表达细胞系中进行了各项功能实验的检测,其中包括通过细胞计数试剂盒8(cell counting kit-8,CCK-8)探究该突变对细胞增殖的影响,并发现RTEL1-MUT的表达会使得细胞的增殖速度变慢. 为进一步了解RTEL1-MUT抑制293细胞增殖的机制,我们通过免疫沉淀以及质谱技术寻找到了与RTEL1的相互作用的蛋白HSP90,并通过Western Blot对相互作用进行了验证,并且发现该突变会导致二者的相互作用减少. 为了深入了解HSP90对RTEL1功能的影响,我们使用HSP90抑制剂STA-9090处理过表达野生型RTEL1(RTEL1-WT)以及突变型RTEL1(RTEL1-MUT)的293细胞系,发现HSP90的活性会影响RTEL1的稳定性.基于以上结果,我们推测该患儿的RTEL1突变可能通过影响细胞增殖而导致先天性角化不良的发生,而RTEL1功能的改变可能源于突变影响了与HSP90的相互作用,从而影响了RTEL1的稳定性.
基于目前的研究结果,依然存在很多问题亟需解决.在后续的研究中,我们首先需要对HSP90通过RTEL1抑制细胞生长进行进一步地明确.并且目前尚少有关于HSP90与RTEL1相互作用的直接报道,仅已有报道表明,HSP90作为伴侣蛋白可以与端粒酶复合物结合并起到维持其活性的作用[20],因此对于HSP90与RTEL1相互作用对RTEL1功能的影响进行研究至关重要.此外,我们目前的研究处于初步探究RTEL1突变导致遗传性骨髓衰竭综合征发生的机制,因此通过工具细胞293细胞来进行功能实验研究.而要更加深入地探究RTEL1突变所带来的疾病相关表型以及导致遗传性骨髓衰竭综合征发病的机制,还需要在更多种类细胞中进行研究.由于DC患者主要的临床表现包括骨髓衰竭、皮肤角化不良以及肺纤维化,因此我们计划后续分别在骨髓CD45+细胞、肺成纤维细胞以及皮肤角质细胞中研究RTEL1突变所造成的表型.除体外实验外,也需要选择合适的动物模型(如斑马鱼)来对RTEL1在疾病发生中所发挥的功能进行进一步的印证.
综上,本研究对于遗传性骨髓衰竭综合征的精准诊断有着重要参考意义,使我们对该病的发病机制有了更加深入的了解,揭示了RTEL1突变导致先天性角化不良发生的机制,揭示了RTEL1起到影响细胞增殖的功能,并且发现了HSP90与RTEL1的相互作用,为后续对RTEL1功能的进一步研究提供了良好的基础.
