4个Currarino综合征家系的临床表型及遗传学分析
2023-01-15刘凌娅耿园园首都儿科研究所遗传研究室北京0000首都儿科研究所附属儿童医院普外科通讯作者mailteacocom
刘凌娅,张 震,李 颀,耿园园,李 龙,姜 茜(首都儿科研究所遗传研究室,北京 0000;首都儿科研究所附属儿童医院普外科;通讯作者,E-mail:teaco@6.com)
Currarino综合征(CS, OMIM# 176450)于1981年首次描述,是一种以骶骨发育不全、肛门直肠畸形和骶前肿物为主要三联征表现的罕见先天性疾病,发病率约为(1~9)/100 000[1,2]。骶骨发育不全常表现为镰刀状或新月状半骶骨,骶前肿物性质多为良性,肛门直肠畸形可表现为肛门闭锁、肛门狭窄和肛门异位等,患者也可伴有其他系统的异常[1,3,4]。研究发现CS临床表型多变,部分患者并不表现为完全的三联征。2004年Martucciello等[5]提出可将CS分为完全型(表现典型三联征)、轻型(骶骨发育不全伴肛门直肠畸形或骶前肿物)及微型(仅有骶骨发育不全)3种[5]。CS患者的诊断常采用多学科联合的方式进行,对以肛门直肠畸形或不明原因慢性便秘为主要表现的患者根据其临床表型特点、家族史、影像学、遗传学检查等进行诊断,并对有可疑症状的亲属完善相关检查以分析是否为家族性病例[4-6]。
遗传学研究提示CS是一种常染色体显性遗传疾病,存在不完全外显、临床表型异质性的特点[7]。MNX1基因(gene ID:3110)杂合突变被认为与CS的发生密切相关[8]。既往研究在几乎所有家族性CS病例和约30%的散发性CS病例中检出MNX1突变[4,9]。MNX1基因位于染色体7q36,有3个外显子,编码一个由401个氨基酸构成转录因子蛋白HB9,包含一个高度保守的同源异形盒区域(第244-297位氨基酸)[3,4,10]。迄今为止已在108例家族性病例及55例散发性病例中检出MNX1基因变异[11],但MNX1参与CS发生的具体机制至今尚未阐明,其上、下游作用因子及可能的参与致病的其他基因仍有待发掘。本次研究通过招募4个家系的CS先证者及其父母,分析所有检出的CS患者的临床表型特点,并通过遗传学检查检测其是否携带MNX1基因致病突变及其他可疑突变,分析其遗传学特征,以寻找可能存在的热点突变及其他可能参与致病的候选基因及其变异。
1 资料与方法
1.1 临床资料
本研究收集从2021年4月到2021年11月期间在首都儿科研究所附属儿童医院普外科就诊并接受治疗的4个CS家系先证者的临床资料,包括病史(确诊年龄、诊疗经过、三联征表现及其他系统症状、家族史等)、影像学检查等明确CS诊断。所有CS家系中先证者父母均非近亲结婚,除家系1中先证者母亲也被确诊为CS外,家系2,3,4中先证者的父母均无类似疾病家族史且兄弟姐妹均体健,收集并分析家系中检出的CS患者的临床表型信息。本研究经过首都儿科研究所伦理委员会批准(SHERLL2019049),所有患者的监护人均已签署知情同意书。
1.2 研究方法
1.2.1 临床表型分析 4个家系中,确诊患者均符合CS的诊断标准,即检出骶骨畸形,同时合并肛门直肠畸形、骶前肿物或其他CS的相关异常。通过收集分析患者家族史,对先证者家庭中有相关病史的亲属完善检查以判断是否为家族性病例。依据2004年Martucciello等[5]提出的分型方法对4个家系中的所有CS患者进行临床分型:①完全型,表现典型三联征,即骶骨发育不全、骶前肿物、肛门直肠畸形;②轻型,骶骨发育不全伴肛门直肠畸形或骶前肿物;③微型,仅表现骶骨发育不全。
1.2.2 目标基因全外显子组测序及突变分析 留取先证者及其父母的外周血样本,使用Miller经典盐析法提取基因组DNA,于北京迈基诺基因科技股份有限公司对患者基因组DNA进行目标基因全外显子组的高通量测序,过滤后的数据与人类基因组(GRCh37/hg19)进行比对,对检出的突变根据美国医学遗传学与基因组学会(American College of Medical Genetics and Genomics, ACMG)遗传变异分类标准与指南[12]进行致病性分析,最终筛选出与临床表型高度相关且致病性证据较为充分的基因变异。同时对检出点突变患者的父母DNA使用Sanger测序法验证以确定变异来源。
1.2.3 文献复习 利用PubMed、dbVar、ClinVar数据库查阅CS相关文献,比较分析与本次检出的点突变位点相同或缺失范围近似的Currarino综合征患者的临床表型特点。
2 结果
2.1 CS患者的临床表型特征
共纳入4个CS家系,收集并分析先证者家族史后,发现家系1中先证者母亲既往有CS相关临床表现,完善检查后也被确诊为CS,其余家系中的先证者父母、兄弟姐妹均未报告CS相关临床症状,因此4个家系中共检出5例CS患者,包括1名男性、4名女性。按照Martucciello等提出的方法进行临床分型,3例患者表现为完全型CS,2例患者表现为轻型CS。所有CS患者均表现骶骨畸形和肛门直肠畸形,除典型三联征外,本次检出患者还伴有神经系统(脊柱裂、脊髓栓系)、泌尿系统(膀胱顺应性降低)、生殖系统(双子宫畸形)及骨骼系统(耻骨及髂骨畸形)中的部分异常表现(见表1)。
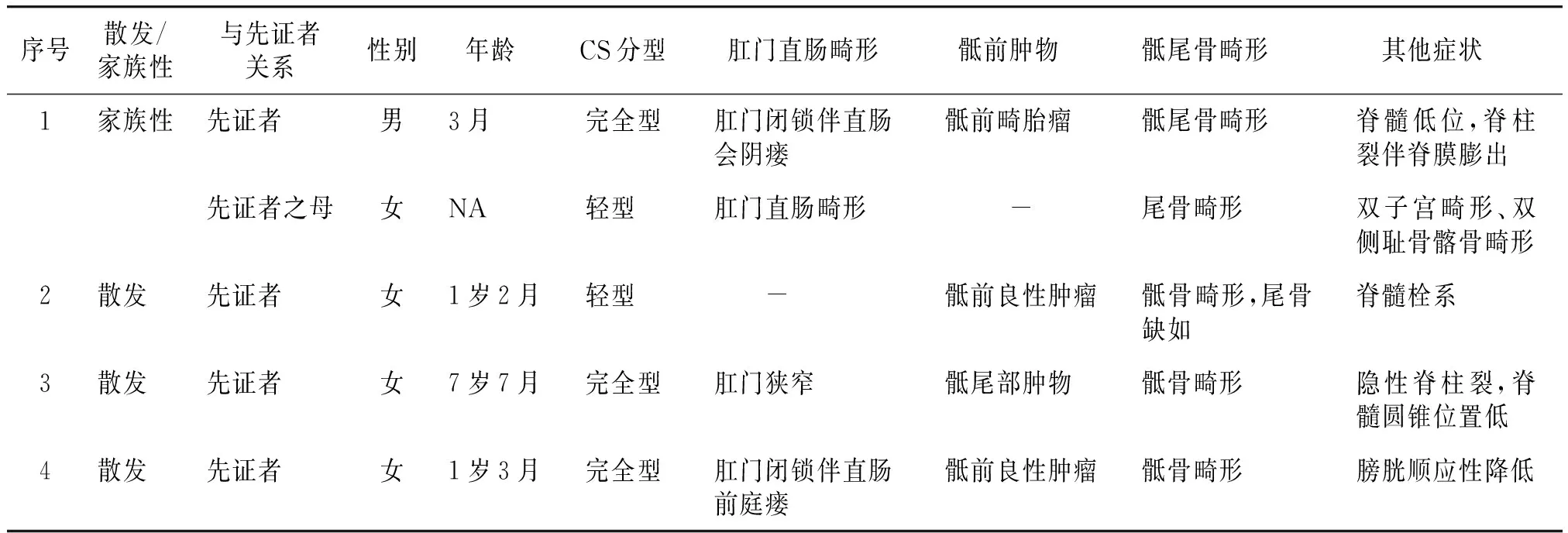
表1 4个家系中5个CS患者的临床表型Table 1 Clinical phenotype of five CS patients in 4 pedigrees
2.2 目标基因全外显子组测序与突变检测
采集上述4个家系中CS先证者及其父母、兄弟姐妹的外周血并提取基因组DNA,共收集13人样本。通过对儿童肠道先天发育异常性疾病已知的目标基因全部外显子进行高通量测序,共检出来自不同家系的3种MNX1基因的致病突变,包括1个错义突变(c.877C>T p.R293W,家系1),1个移码突变(c.53dupC p.R19Tfs*37,家系2)和1个拷贝数缺失(家系3,位于7q36.3,约0.219 Mb,缺失区域内包括MNX1基因和LMBR1基因),全部为杂合突变。3种突变位点的分布模式见图1。
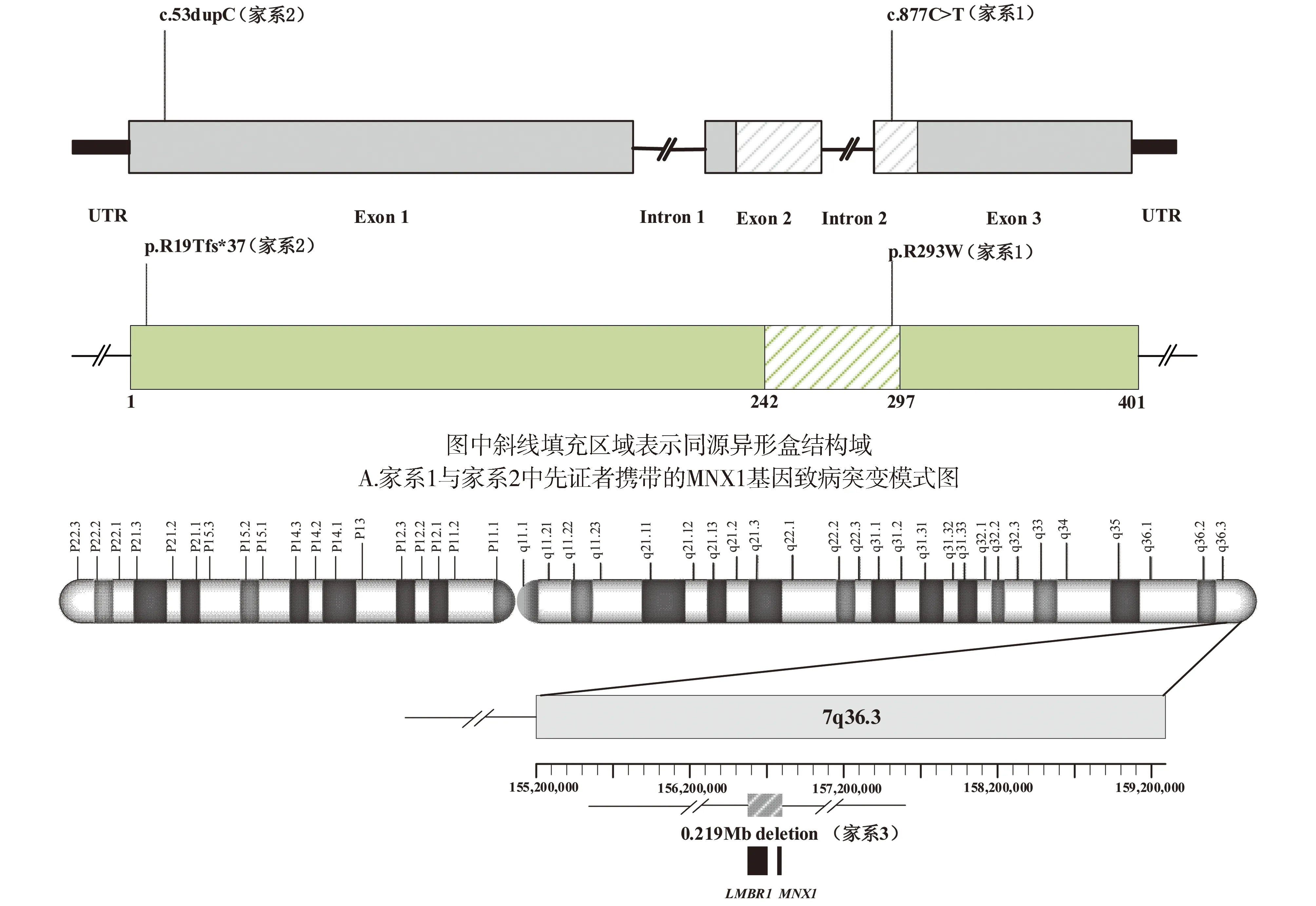
灰色斜线区域表示7q36.3(区域:chr7:156583817-156803063)0.219 Mb缺失范围,黑色填充框表示该缺失范围内包含的基因B.家系3中先证者携带的染色体7q36.3区域拷贝数缺失示意图(GRCh37/hg19)图1 CS患者突变位点模式图Figure 1 Schematic representation of the mutation sites in CS patients
4个家系中所有CS患者的信息及其携带MNX1基因致病突变的测序结果见图2、图3。家系1中,先证者及其母亲被确诊为CS并均携带1个位于MNX1基因第3外显子的杂合错义突变c.877C>T(p.R293W),先证者父亲未检出突变且无CS相关临床表现(见图2)。该突变位点编码的氨基酸位于高度保守的同源异形盒结构域(见图1A),曾在既往CS病例中被报道[13],根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics, ACMG)遗传变异分类标准与指南评估为致病性变异(PS4+PM1+PM2+PM5+BP4)(见表2)。

家系图中黑色填充图形表示CS确诊患者,(+)表示携带MXN1致病突变(具体突变位点及缺失区域在家系图下方注明),(-)表示未检出MNX1致病突变图2 CS家系1、家系2的家系图及测序结果Figure 2 Pedigree and sequencing results of the pedigree 1 and pedigree 2 with CS
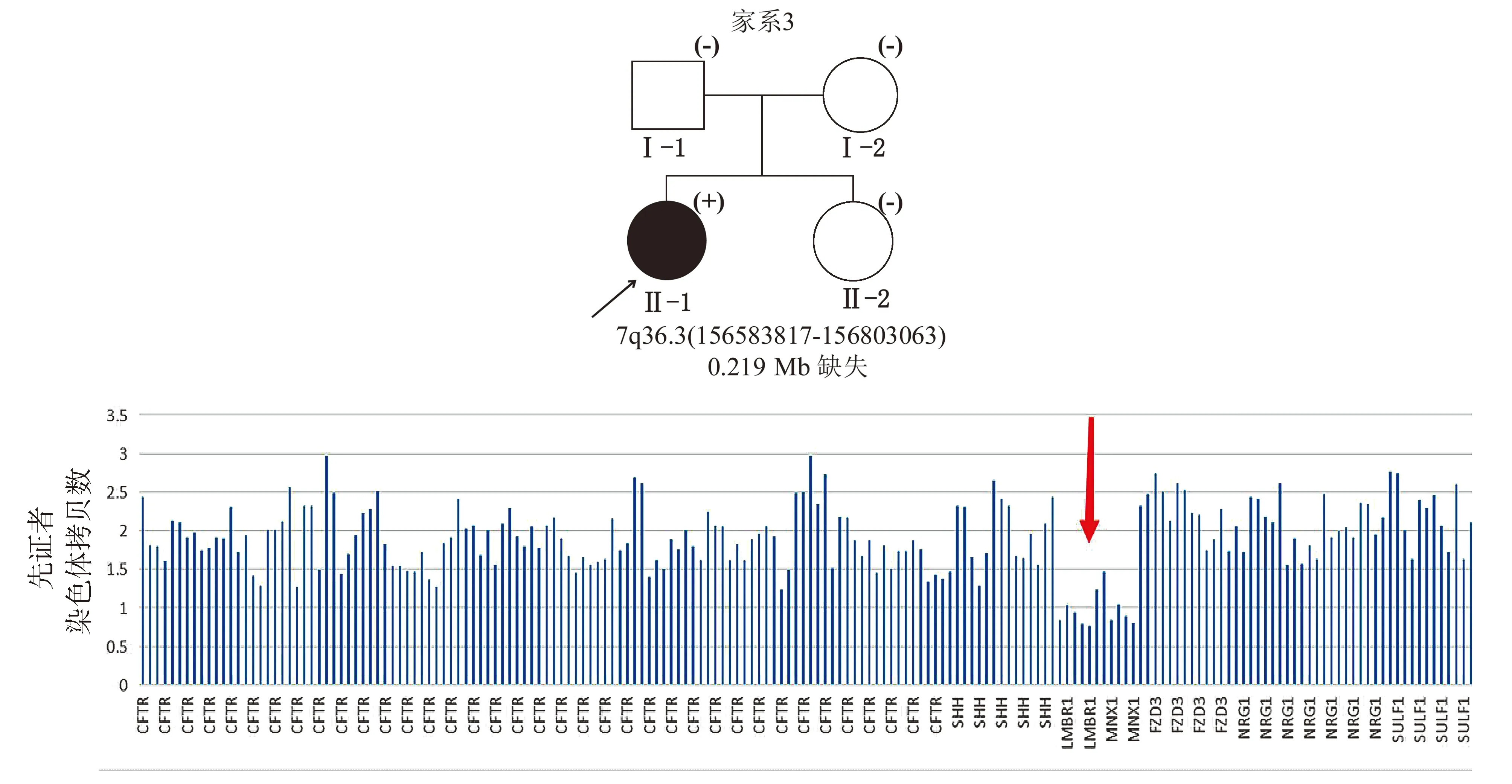
黑色填充图形表示CS确诊患者,(+)表示携带MXN1致病突变,(-)表示未检出MNX1致病突变;红色箭头指示拷贝数缺失区域,该区域累及MNX1、LMBR1基因图3 CS家系3的家系图及测序结果Figure 3 Pedigree and sequencing results of the pedigree 3 with CS
家系2中,高通量测序结果显示先证者携带1个位于MNX1基因第1外显子的杂合移码突变c.53dupC(p.R19Tfs*37),变异来源自先证者父亲,但先证者父母均无CS相关临床表现(见图2)。该突变在既往文献中曾8次报道于不相关的CS患者[4,7,14-17],确定为致病性变异(PVS1+PS4)(见表2)。

表2 CS家系先证者遗传学检测结果Table 2 Genetic results of the CS probands
家系3中,先证者携带1个位于染色体7q36.3(区域:chr7:156583817-156803063)的拷贝数缺失,为大小约0.219 Mb的杂合缺失(见图3),该区域内包含MNX1基因及LMBR1基因,为致病性变异,先证者父母及其妹妹均未检出该致病突变,为新发突变(见表2)。先证者父母及妹妹均无CS相关临床表现。dbVar数据库中(https://www.ncbi.nlm.nih.gov/dbvar/)已有相似区域拷贝数缺失的CS病例报道(病历号:nsv997030|区域:chr7:156797547-156803347|Pathogenic|病例临床表型:Currarino综合征),具体表型不详。
家系4中,先证者经高通量测序未检出MNX1基因的致病突变,但携带1个位于SHH基因的杂合错义突变c.488G>A(p.R163H)(见图4),变异来源为先证者母亲,先证者父母均无CS相关临床表现。SHH基因突变曾在多个CS病例中报道[18,19]。
2.3 文献回顾
我们回顾了既往报道中与本研究CS患者中检出的MNX1基因致病点突变的基因型相同或缺失范围近似的CS患者的临床表型,并对散发性病例及家族性病例中的先证者临床表型进行分型(见表3)。本次检出的错义突变曾在一个CS家族性病例中被报道,但该文献未详述先证者及家系中其他患者的突变携带情况及对应表型信息,故无法进行临床分型。本次检出的移码突变曾在多达8个CS病例中被报道,除1例临床表型信息不明外,其他7名患者的临床分型均为完全型CS。家系3中检出的拷贝数缺失,dbVar数据库曾报道1例携带近似区域拷贝数缺失的CS患者,但临床表型未明。
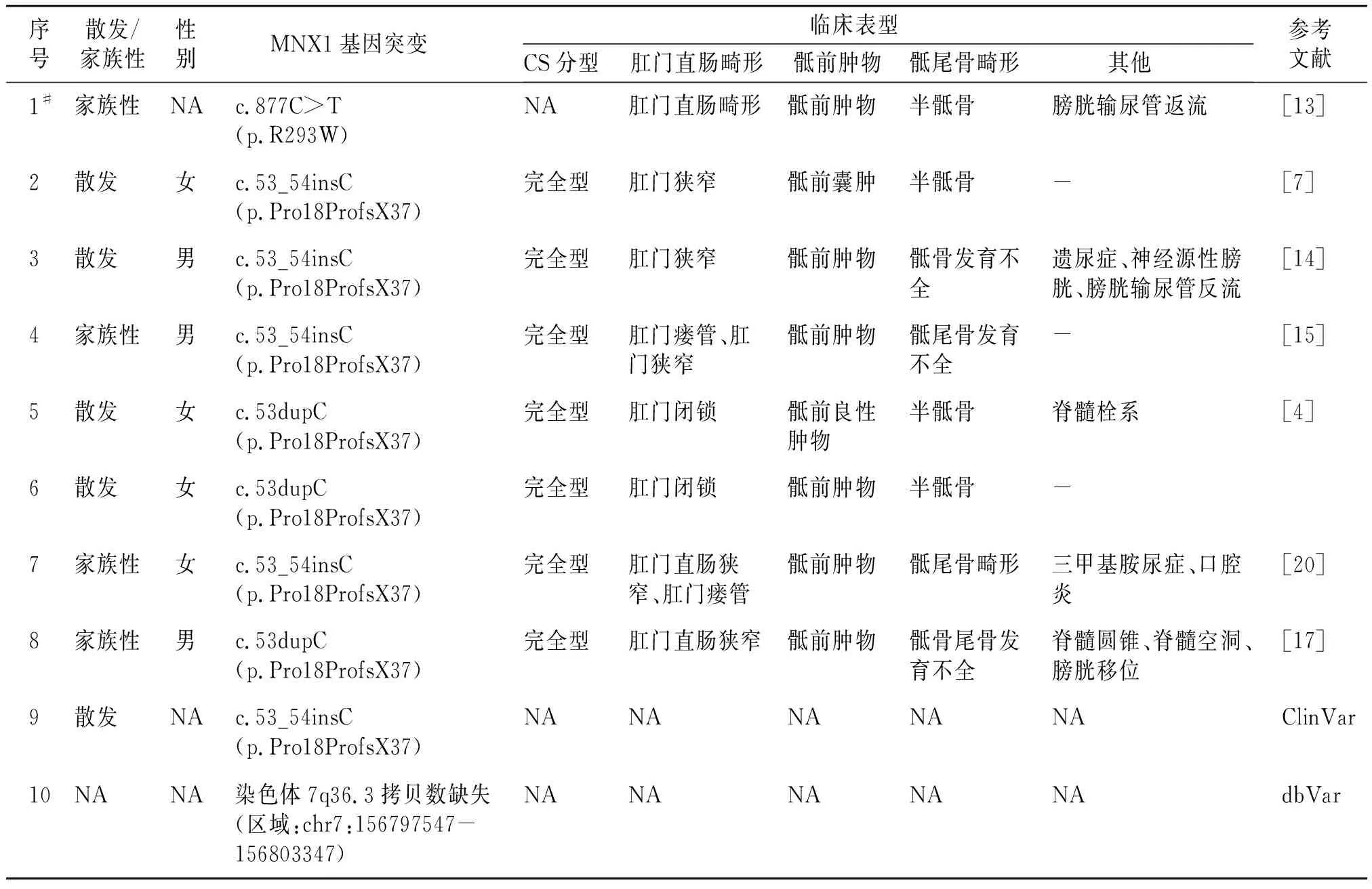
表3 既往携带相同点突变或近似缺失范围的CS患者基因型与表型信息Table 3 Genotype and phenotype information of previously reported CS patients carrying same point mutations or similar deletions
3 讨论
3.1 CS患者的临床表型特征
本研究围绕4个CS家系进行,其中3个散发性病例,1个家族性病例,共检出5名CS患者,临床分型2人为轻型CS,3人为完全型CS(见表1)。除典型的三联征外,5例患者还分别伴有神经系统、泌尿系统、骨骼系统及生殖系统等其他系统的部分异常表现,其中3例患者有神经系统的异常表现并均携带MNX1致病突变(见表1),包括脊柱裂伴或不伴脊膜膨出、脊髓低位、脊髓圆锥低位和脊髓栓系,这可能提示携带MNX1突变的CS患者中神经系统受累并不罕见。2008年Crétolle等[4]在50例CS患者中发现约70%携带MNX1基因突变者有神经管畸形的表现,其中脊髓栓系、脊髓圆锥低位和椎管内脂肪瘤较多见,与本研究结果相似,因此也有研究者认为神经系统畸形应被列为CS的第四大临床表型特征。
值得注意的是,本次检出的家族性病例中先证者母亲表现双子宫畸形,回顾既往CS病例发现CS患者可伴有双子宫畸形、双角子宫、阴道隔等多种女性生殖系统畸形[7,21]。有研究认为这些生殖系统畸形与苗勒氏管发育畸形(Müllerian duct anomalies, MDAs)有关,CS患者合并MDAs的发生率高达15%[9,22],而普通女性人群中MDAs患病率约为6.7%[23],提示CS女性患者有更高风险患生殖系统畸形疾病,应在临床诊疗过程予以关注。
本研究中CS患者男女比例为1 ∶4,但限于样本量较小,尚不足以说明男女比例存在统计学差异。既往研究提示CS患者中男女患者比例约为1 ∶1.39,可能与CS女性患者MDAs的发生率较高有关[4]。
3.2 基因型-表型关联
目前研究认为MNX1为CS的致病基因,本研究4个家系中共检出3种MNX1基因致病突变,包括2个点突变及1个累及MNX1基因的拷贝数缺失。携带相同点突变或近似缺失的CS病例在既往研究中已有报道[4,7,13-17]。家系1检出的MNX1错义突变c.877C>T(p.R293W)曾在1个临床表现为肛门直肠畸形、半骶骨畸形、骶前肿物及输尿管膀胱返流的CS家族性病例中报道(见表3),但先证者临床表型未明,故无法进行临床分型[13]。该错义突变位于MNX1基因编码蛋白中高度保守的同源异形盒区域内,既往研究表明该区域参与HB9蛋白与“TAAT”DNA基序靶标的结合,或通过体外核定位信号(NLS)基序参与其核定位,因此该区域的突变可能影响DNA结合特异性或影响突变蛋白的核定位[10,24]。既往CS患者检出位于同源异形盒区域内的突变较为常见,该功能域参与疾病发生、发展的过程及机制值得进一步研究[4,21]。
家系2携带的MNX1基因移码突变c.53dupC(p.R19Tfs*37)曾在8个互不相关的CS散发性患者及家族性病例中被报道,除1例先证者的临床表型不明外,其他先证者均为完全型CS(见表3)[4,7,14-17],而本研究中家系2先证者的临床分型为轻型CS,即病例7/8携带该突变且临床表型明确的CS先证者为完全型CS,存在显著表型异质性。
家系3检出1个累及MNX1基因与LMBR1基因的拷贝数缺失,携带相似区域拷贝数缺失的CS病例在dbVar数据库中已有报道,但其临床表型未明。早在1996年就有研究人员指出部分CS患者的染色体7q36区域发生杂合缺失[25],后续研究提示这些染色体缺失往往不仅累及MNX1基因,还累及SHH等位于染色体7q36上的其他基因,并被认为可能与CS患者的一些表型相关,如SHH基因被认为可能和部分CS患者的小头畸形等表现有关[4,18,19]。而本次累及的LMBR1基因变异曾在2个不相关的携带MNX1突变的CS病例中被报道,目前LMBR1是否参与CS发生尚未明确[26]。
家系4先证者虽未检出MNX1基因致病突变,但检出一个SHH基因的错义突变c.488G>A(p.R163H)。既往有研究分析10例携带染色体7q36.3区域缺失的CS患者临床表型,发现5例患者有不同程度CS临床表现,认为SHH基因可能参与CS的发生[8,18]。SHH是表达最广泛的哺乳动物HH信号分子,在中胚层分化中发挥重要作用,并参与调控胚胎骶骨发育、神经管发育、人类胚胎期肠道发育及区域特化等多个过程[27]。目前研究对SHH是否参与CS的发生尚无定论,也有研究者认为CS的发生可能与SHH无关[28]。该突变的检出提示对CS致病机制的进一步研究或可从SHH基因入手,重点关注该基因及SHH通路中的重要分子是否参与CS的发生发展。
CS患者临床表型复杂多变,即使在家族性病例中携带相同MNX1致病突变的个体间临床表型亦不相同,提示可能有其他基因或调节元件直接或间接参与疾病发生,未来研究应重视并收集CS患者中多次检出的非MNX1基因变异并分析、评估其是否参与CS致病,以寻找新的候选基因。
综上,CS患者临床表型变异大,部分患者经多次诊疗后才得以确诊,分析CS的临床表型与基因型特征,探讨CS家系中需要关注的临床表现(如三联征、神经系统或女性患者生殖系统的异常等)及MNX1基因突变特点,对帮助更多患者明确诊断、检出家系中其他携带者、提供遗传咨询具有重要意义。尽管MNX1基因在CS中的致病机制尚不明确,其上、下游作用因子仍有待研究,携带热点突变位点的患者得到确诊可能提示新的研究思路,同时也应重视收集在多个不相关病例中反复检出的非MNX1基因变异并进行遗传学分析以寻找新的候选基因。目前随着对CS这一罕见病的认识逐渐提高、诊疗技术的进步及遗传学检测的大规模开展,更多的CS患者得以确诊,未来随着更多临床病例的收集及遗传学的深入研究有望最终明确其发生机制,为临床诊疗提供更多帮助。
