伪狂犬病病毒IE180基因启动子缺失克隆的构建与鉴定
2021-05-20韩冬梅阮可悦曹云雷张丽荣李国新童光志
韩冬梅,阮可悦,曹云雷,张丽荣,童 武,李国新,郑 浩,,童光志,
(1.中国农业科学院上海兽医研究所,上海200241;2.扬州大学 江苏省动物重要疫病与人兽共患病防控协同创新中心,扬州225009)
长期以来,基因缺失突变株的构建是研究疱疹病毒功能基因的主要策略。1997年,Messerle等[1]首次将细菌人工染色体(BAC)应用于疱疹病毒,成功构建了巨细胞病毒(Cytomegalovirus,CMV)感染性细菌人工染色体克隆。该技术已成为疱疹病毒分子病原学、基因功能研究不可或缺的工具。本研究在伪狂犬病病毒变异毒株JS-2012的感染性克隆pBAC-JS2012及突变筛选菌株SW102-PRVBAC基础上进行IE180启动子的缺失[2]。
伪狂犬病(pseudorabies, PR或Aujeszky’s disease,AD)是一种由伪狂犬病病毒(Pseudorabies virus,PRV)感染引起的多种家畜和野生动物共患的急性传染病。猪是伪狂犬病病毒的唯一宿主,猪伪狂犬病的暴发和流行给全世界养猪业造成了巨大的经济损失[3-4]。PRV基因组全长约145 Kb,GC含量高达74%左右,包含70多种基因,可编码100多种蛋白,值得注意的是PRV与其他疱疹病毒相比只有一个立即早期基因即IE180与单纯疱疹病毒ICP4基因同源,其在基因组中存在两个拷贝,且大部分序列与病毒潜伏感染相关转录本(large latency transcripts,LLT)重叠,是病毒复制和早期基因转录所必需的。
根据PRV感染后基因转录的时间顺序可将其分为三类:立即早期基因(immediate-early gene,IE)、早期基因(early gene,E)、晚期基因(late gene,L)。IE基因的转录出现在病毒DNA复制之前。IE基因转录后,激活早期基因和晚期基因的表达,引起病毒裂解性感染。IE180是PRV唯一的一个立即早期基因,它的表达不需要任何病毒蛋白的合成,IE180是整个基因表达的反式作用因子[5-7]。IE180启动子改变后的重组PRV不感染细胞,证明IE180的正常诱导表达与病毒能否感染培养细胞有关[8]。将PRV IE180基因两个拷贝同时缺失,重组病毒不复制,不能产生子代病毒[9]。虽然IE180影响着细胞基因的表达,但IE180基因缺失的重组PRV仍能与稳定表达IE180蛋白的Vero和PK-15细胞系存在着互补效应,同时细胞系中IE180基因的表达对内源逆转录病毒的增殖起到了强化作用[10],同时IE180蛋白对细胞基因的影响仍需更进一步研究。
1 材料与方法
1.1 病毒、细胞、质粒和主要酶与试剂 伪狂犬病病毒变异株JS-2012感染性细菌人工染色体pJS-2012BAC插入SW102菌株的阳性克隆SW102-PRVBAC、野毒株JS-2012、Vero细胞、BHK-21细胞、IE180多克隆抗体、pCAGGS-MCS载体、pCR-Blunt Ⅱ TOPO载体和pgalk载体均由本实验室提供。限制性内切酶、T4 DNA连接酶、大肠杆菌DH5α、Oligo dT反转录慢酶、Lipofectamine3000转染试剂、Prime STAR Max DNA等PCR相关试剂购自TaKaRa公司;胎牛血清、DMEM购自Gibco公司;Trizol(总RNA抽提试剂)、氨苄青霉素和氯霉素购买自Invitrogen公司;20%半乳糖、5×M63和1×M9购自生工生物工程(上海)有限公司;D-生物素购自上海联硕生物科技有限公司;亮氨酸(Leucine)购自上海吉尔生化科技有限公司;SOC培养基购自上海前尘生物科技有限公司;凝胶回收试剂盒购自广州东盛生物科技有限公司。
1.2 培养基及常用溶液的配制 氯霉素储存液:500 mg氯霉素溶于20 mL无水乙醇中,混匀后于超净台中0.22 µm滤膜过滤除菌,-30℃保存备用。卡那霉素储存液:500 mg卡那霉素溶于20 mL灭菌ddH2O中,混匀后于超净台中0.22 µm滤膜过滤除菌,-30℃保存备用。M63溶液:(5×)2.5 mg七水硫酸铁、10 g硫酸铵、68 g磷酸二氢钾和8.58 g M63颗粒溶于110 mL ddH2O并调pH为7.0,高压灭菌后,4℃保存备用。M9溶液:(1×)0.5 g氯化钠、3 g磷酸二氢钾、6 g磷酸氢钠、1 g氯化氨和0.565 g M9颗粒溶于50 mL ddH2O中,高压灭菌后,4℃保存备用。20%Galactose:1 g Galactose溶解于9 mL去离子水中,定容至10 mL,高压灭菌后,4℃保存备用。D-生物素(避光):25 mL去离子水中融有5 mg D-生物素,0.22 µm滤膜过滤除菌,4℃冰箱保存备用。10%细胞生长液:450 mL DMEM中加入50 mL FBS与2 mL青链霉素双抗,混匀,4℃冰箱保存备用。2%细胞维持液:500 mL细胞培养基中加入10 mL胎牛血清和2 mL青链霉素双抗,混匀,4℃冰箱保存备用。LB液体培养基:10 g Tryptone、5 g Yeast Extract和10 g NaCl溶于800 mL ddH2O中,调pH为7.0,定容到1 L,高压灭菌后加入相应抗生素,4℃保存备用。
麦康凯培养基:麦康凯琼脂粉10 g溶于200 mL去离子水中,高压灭菌后冷却至50℃左右加2 mL 20%Galactose和100 µL氯霉素,倒板备用。LB固定培养基:在LB液体培养基中加入15 g琼脂粉,其余相同(倒板前冷却至50℃左右时加抗生素)。galk正筛选培养基:高压灭菌后的液体琼脂(7.5 g琼脂溶于0.8 L ddH2O),加入100 mL高压灭菌后的5×M63溶液,定容到0.5 L。待冷却至50℃左右加入2.25 mL L-亮氨酸、5 mL 20%Galactose、2.5 mL生物素、250 µL氯霉素倒平板备用。galk二次正筛选固体培养基:同galk正筛选培养基外,补加卡那霉素250 µL。
1.3 引物的设计与合成 根据要删除的PRVJS-2012 IE180启动子基因序列,设计扩增galk和Kna表达盒及两端带有50 bp同源臂的引物。所有引物均由上海英潍捷基有限公司合成,序列见表1。

表1 引物及序列Table 1 Primers and sequences used in this study
1.4 正筛选技术路线 正筛选:借助同源重组的方法,将galk基因插入并替换pJS-2012BAC的IE180基因启动子及其两端200 bp基因序列,生长于正筛选培养基上的菌落,基本上全是galk表达盒成功替换掉缺失部位的重组子菌落。正二筛:在首次筛选的基础上,操作方法相同。将Kna抗性基因代替galk表达盒,替换pJS-2012BAC的另一个IE180基因启动子序列。经BamHⅠ酶切的长度多态性分析和PCR鉴定,双拷贝启动子缺失后全基因组序列没有大片段的插入与缺失可获得pJS-2012BAC的DIE180启动子基因的克隆,正筛选构建PRV IE180启动子缺失病毒过程示意图见图1,IE180启动子缺失位置分别在两个基因拷贝的不同序列位置,其中一个缺失位置在反向拷贝启动子两端的左侧297 bp和右侧172 bp,而另外一个缺失位置在正向拷贝的启动子两端,即左侧134 bp和右侧309 bp。详细示意图见图2。
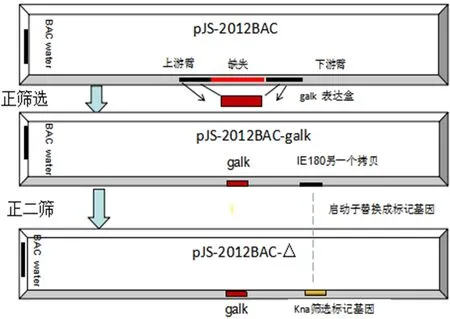
图1 构建PRV IE180缺失病毒的正筛选示意图Fig.1 Generation of IE180 promoter deleted PRV using the galk selection system and pJS-2012BAC
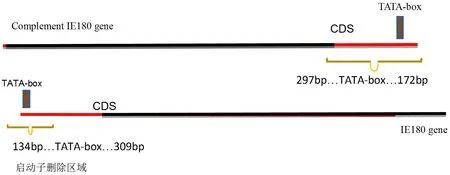
图2 IE180启动子缺失位置示意图Fig.2 The schematic of IE180 promoter missing position
1.5 IE180两个拷贝缺失克隆构建中PCR扩增片段基因galk及Kna表达盒 分别用引物galkup/galkdown和Knaup/Knadown各1 µL以pgalk和pCR-Blunt Ⅱ TOPO质粒为模板扩增片段基因galk及Kna表达盒。PCR扩增体系(50 µL):2× buffer 25 µL,上、下游引物(10 μmol/L)各1 µL;2.5 mmol/L dNTP 4 µL;Primer star高保真DNA聚合酶0.5 μL;质粒模板2 µL;ddH2O 16.5 µL。Kna和galk基因PCR扩增反应程序:95℃预变性5 min;95℃变性30 s;60℃退火30 s;72℃延伸1 min,共35个循环;72℃再延伸10 min。PCR结束后,PCR产物加入2 µL DpnⅠ37℃酶切1 h,对PCR产物进行核酸凝胶电泳分析,观察扩增情况。胶回收目的片段,4℃保存备用。
1.6 IE180两个拷贝缺失克隆构建的正筛选 分别用引物galkup/galkdown和Knaup/ Knadown扩增Galk和Kna的带有缺失部分两边同源臂的筛选标记基因。用图1中的技术路线将IE180缺失部分进行替换。(1)和(2)操作步骤同文中正筛选[11]。(1)SW102-PRVBAC正筛选电转感受态的制备。(2)正筛选的电转化。(3)用麦康凯培养基对筛选到的正筛阳性克隆进行纯化:挑取正筛选平板上的单菌落接种于麦康凯培养基,使用跨galk引物Lgalkup/Lgalkdown鉴定galk正确插入,PCR扩增体系与扩增程序同1.5。将正确插入galk的正筛阳性菌株提取质粒后命名为pJS-2012BAC-galk保存于-20℃。本过程中所有的菌体培养模式均为避光培养且温度不高于32℃。(4)IE180第二次正筛选(缺失IE180另一个拷贝启动子):在IE180一个拷贝启动子缺失克隆SW102-IE180-galk的条件下进行另一个拷贝启动子的缺失,操作步骤如上3.2.4中的(1)、(2)和(3),只是将过程中所需的抗性及筛选标记基因进行替换。将Kna成功插入靶向位置并测序正确的阳性克隆质粒命名为pJS-2012BAC-DIE180。
1.7 成功构建IE180双启动子缺失克隆的鉴定
1.7.1 RFLP长度多态性分析 将质粒pJS-2012BAC、pJS-2012BAC-galk和pJS-2012BAC-DIE180进行BamH Ⅰ单酶切,并进行核酸电泳检测。具体的操作步骤如下:将质粒pJS-2012BAC、pJS-2012BAC-galk和pJS-2012BAC-DIE180用BamHⅠ限制内切酶于37℃水浴条件下酶切6 h,体系为:BamHⅠ限制性内切酶3 µL,10×CutSmart Buffer 2.5 µL,质粒pJS-2012BAC、pJS-2012BAC-galk和pJS-2012BAC-DIE180各4 µg,ddH2O补至25 µL。
1.7.2 Western blot分析及荧光表达 将质粒pJS-2012BAC、pJS-2012BAC-galk和 pJS-2012BACDIE180各2 µg转染BHK-21细胞,转染6 h后将转染后的上清液换为含2%FBS的DMEM培养基,继续于37℃ CO2培养箱中培养36 h,观察绿色荧光表达情况。随后对转染后的细胞进行裂解收样和SDS-PAGE电泳分析,IE180多克隆抗体作为一抗(1∶500稀释),HRP标记的羊抗鼠IgG作为二抗(1∶5000稀释),显影后观察结果。
2 结果
2.1 IE180两个拷贝缺失克隆构建中galk及Kna表达盒PCR片段基因扩增结果 分别用引物galkup/galkdown和Knaup/Knadown扩增基因片段galk和Kna,扩增大小分别为1300 bp和1000 bp,与实际大小相符合(图3),并且测序结果与目的序列一致。
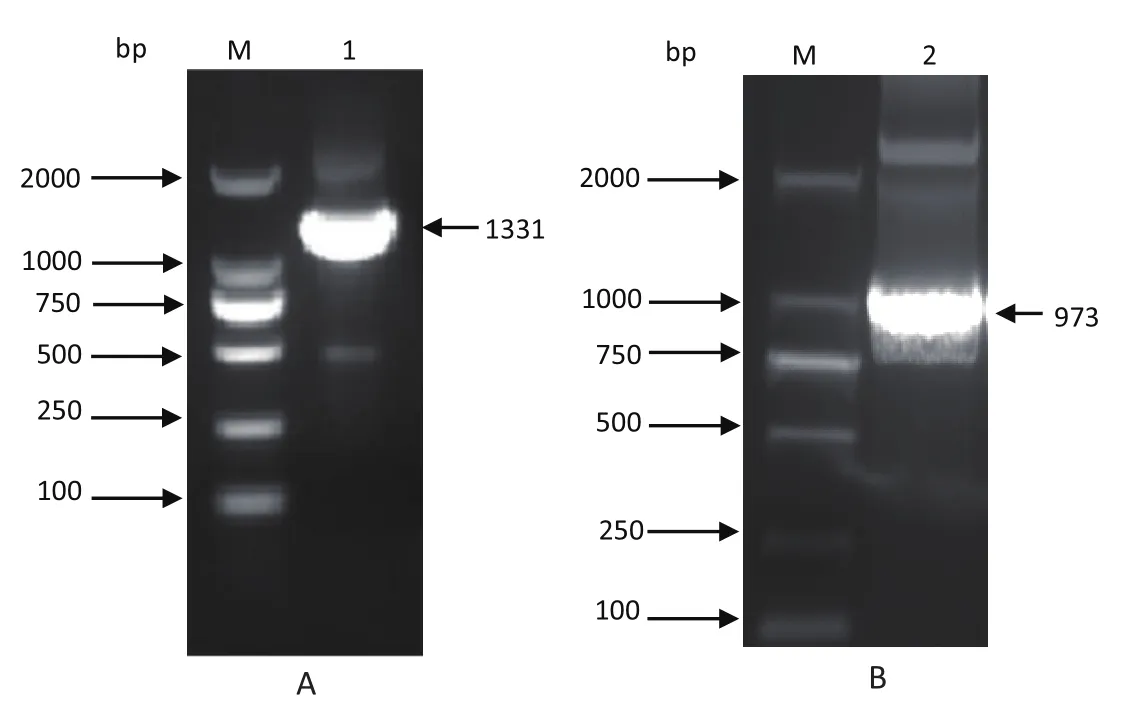
图3 质粒扩增galk(A)和Kna(B)表达盒的核酸电泳图Fig.3 Electrophoresis of amplified galk (A) and Kna (B)fragment
2.2 IE180缺失克隆的两次正筛选鉴定 第一次正筛选(将galk基因表达盒替换IE180基因启动子缺失部分)后用Lgalkup/Lgalkdown进行PCR鉴定,鉴定galk表达盒是否成功替换IE180启动子缺失部分,鉴定长度为842 bp,与实际大小相符(图4)。PCR鉴定后扩增序列经过测序,与实际序列相吻合(图5)。将第一次正筛选得到的阳性克隆用跨IE180启动子缺失的引物IE180up/down进行PCR鉴定,鉴定长度为1260 bp,与实际大小相符合(图4)。由图4的A和B得知IE180另外一个拷贝启动子依然存在,在此基础上进行Kna的二次正筛选删除另一拷贝IE80启动子。用Kna表达盒代替上述galk表达盒进行二次正筛选,筛选PCR鉴定长度为772 bp,与预期大小相同(图4),对二次正筛选的阳性克隆用引物IE180up/down进行PCR鉴定,鉴定IE180两个拷贝启动子是否均删除,与阳性对照(pJS-2012BAC)相比,正筛阳性克隆(pJS-2012BAC-DIE180)无扩增条带(图4)。经二次正筛而筛选到阳性克隆经测序,鉴定Kna筛选标记基因插入位置与预期位置相同(图6)。
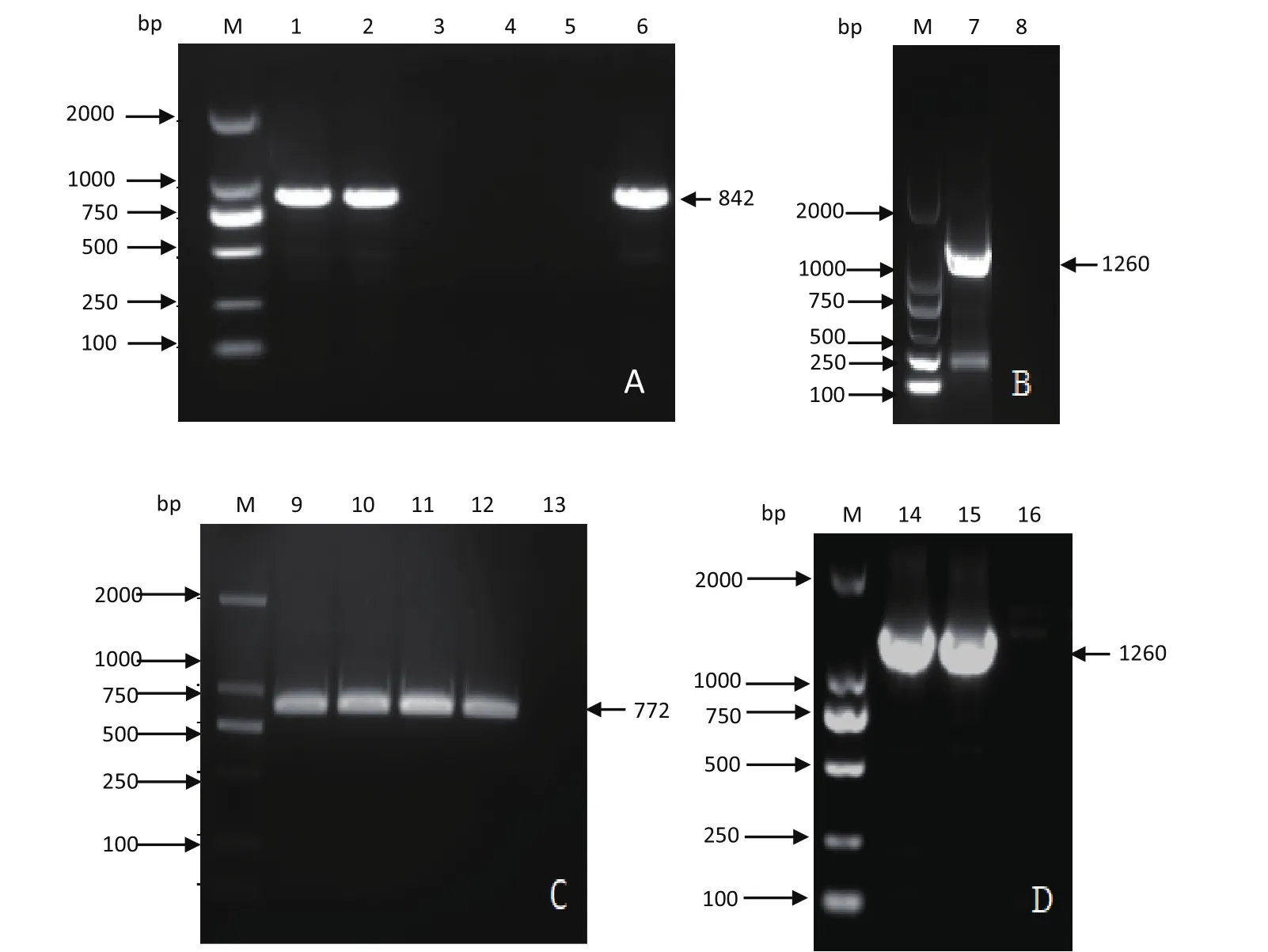
图4 IE180缺失克隆正筛选PCR鉴定Fig.4 IE180 deletion clone positive screening PCR identification
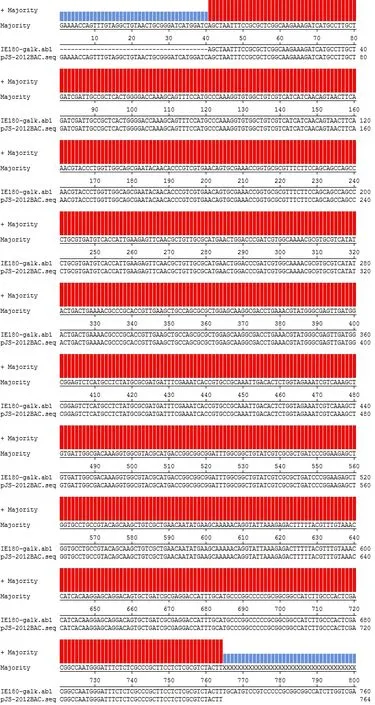
图5 比对PRVJS-2012和pJS-2012BAC-galk测序结果Fig.5 Comparison of sequencing results between PRVJS-2012 and pJS-2012BAC-galk site
2.3 鉴定IE180双启动子缺失克隆的成功构建 利用RFLP长度多态性分析质粒pJS-2012BAC、pJS-2012BAC-galk和pJS-2012BAC-DIE180经BamHⅠ酶切后的片段差异:将质粒pJS-2012BAC、pJS-2012BAC-galk和pJS-2012BAC-DIE180用BamHⅠ限制性内切酶进行酶切,经0.8%琼脂糖核酸电泳分析,发现3个泳道的条带数量和大小无差异,与预期结果相符合(图7)。经过DNAStar软件分析后,启动子删除序列、galk基因序列和Kna基因序列内均无BamHⅠ的酶切位点,所以核酸电泳后3个质粒基因序列的酶切长度无差异。利用p J S-2 0 1 2 B A C、p J S-2 0 1 2 B A C-g a l k和pJS-2012BAC-DIE180质粒转染BHK-21细胞,观察转染后绿色荧光表达情况。3种质粒转染细胞30 h后,观察到pJS-2012BAC和pJS-2012BAC-galk均有绿色荧光表达,但pJS-2012BAC-galk相对于pJS-2012BAC绿色荧光少;pJS-2012BAC- DIE180无绿色荧光表达(图8)。
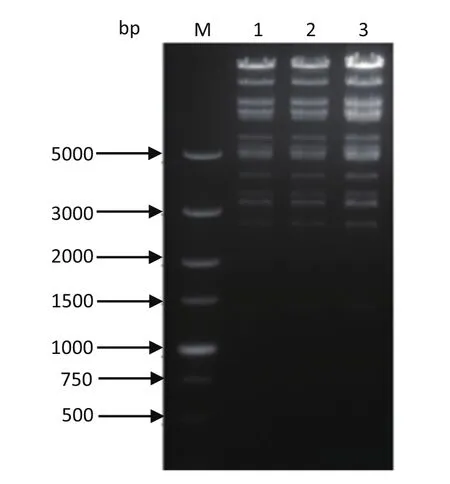
图7 SW102-DIE180 RFLP分析结果Fig.7 RFLP analysis results of SW102-DIE180
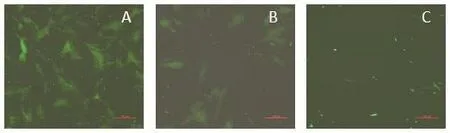
图8 质粒转染BHK-21 细胞Fig.8 Transfection results of the plasmids in BHK-21 cells
利用IE180多克隆抗体检测pJS-2012BAC、pJS-2012BAC-galk和pJS-2012BAC-DIE180 3个质粒IE180蛋白的表达情况:将质粒pJS-2012BAC、pJS-2012BAC-galk和pJS-2012BAC-DIE180各2 µg转染BHK-21细胞,24 h后进行收样,进行SDS-PAGE电泳分析,发现pJS-2012BAC和pJS-2012BAC-galk均有IE180蛋白的表达,而pJS-2012BAC-DIE180无IE180蛋白的表达情况(图9)。
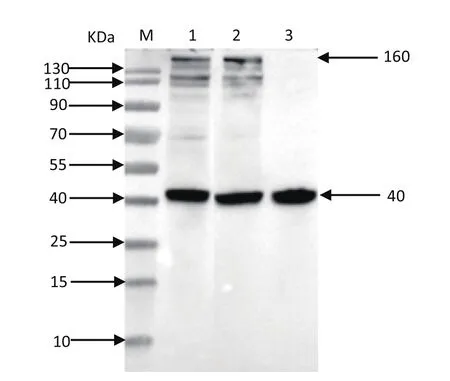
图9 Western blot分析IE180蛋白的表达Fig.9 The expression of IE180 by Western blot analysis
3 讨论
伪狂犬病病毒(PRV)自出现以来给我国养猪业造成极大经济损失,因该病存在着潜伏期难以达到净化与根除而成为养猪业非常重要的一种疾病,目前对于PRV的研究日渐增多,但对于其防控却仍面临着许多挑战[12-13]。
本研究成功构建了将IE180双拷贝启动子删除的含BAC DNA的重组质粒pJS-2012BAC-DIE180。本操作方法简单、快速、高效[14]。在首次正筛选插入galk之后利用同样的方法插入Kna筛选标记基因,Kna是菌株的筛选标记,因而不会对细胞产生影响[15]。利用BAC反向遗传操作和galk正筛选系统缺失PRV IE180基因启动子,经过PCR鉴定发现启动子删除部分仍然存在,由此得知此方法只能将IE180其中一个拷贝的启动子删除,所以在上述方法的基础上在第二个拷贝启动子删除部分插入Kna筛选标记基因替换掉要删除的启动子部分。不同的筛选标记基因Kna和galk同源重组替换掉IE180两个拷贝启动子删除部分,经特异性引物的PCR鉴定和测序结果证明两个拷贝的启动子成功删除。前期研究已经表明,IE180是驱动PRV其他基因转录复制的必需基因,如果IE180不能表达,则整个病毒丧失感染能力,病毒基因组不能进行复制,不能产生子代病毒。本研究将IE180启动子缺失克隆pJS-2012-DIE180和质粒pJS-2012-galk、pJS-2012BAC分别转染BHK-21细胞,经过免疫荧光实验和Western blot分析均表明pJS-2012DIE180转染BHK-21细胞后未表达IE180蛋白;而pJS-2012-galk和pJS-2012BAC绿色荧光均正常表达,但pJS-2012-galk相对于pJS-2012BAC表达量低;pJS-2012-galk和pJS-2012BAC的IE180蛋白正常表达。结果证实IE180启动子缺失克隆构建成功,为进一步构建IE180互补缺失系统及生物学功能的探究奠定理论基础。
IE180作为PRV唯一的一个立即早期基因,在其启动子缺失的情况下,IE180不表达且不能引起PRV整个基因组的复制和转录。IE180双启动子缺失克隆的成功构建,为后期研究IE180互补缺失系统及探寻PRV基因组中与IE180调控机制相关的表达分子提供工具,并且为进一步探究IE180的功能做理论基础。
