混合型肝豆状核变性1例报告
2021-03-08张硕,朱辉,吕洋
张 硕, 朱 辉, 吕 洋
肝豆状核变性(hepatolenticular degeneration,HLD)又称为Wilson病(Wilson’s disease,WD),是一种常染色体单基因隐性遗传的铜代谢障碍性疾病,由于编码铜跨膜转运蛋白的基因ATP7B发生突变,以致铜在肝脏、大脑及其他组织器官中过度沉积。多数HLD患者以肝病和神经精神症状起病,常累及多个器官,最初的症状往往不易与其他神经系统疾病相鉴别,易误诊[1]。现报道我科收治的1例临床上较少见的以运动功能障碍为首发症状的混合型肝豆状核变性患者,以提高对该病的认识。
1 临床资料
患者,男,28岁,因“言语不清伴行走不稳13 y,加重伴肢体扭转9 m”于2019年12月入院。患者于入院前13 y家人发现其讲话不如以往清晰、流利,同时出现行走缓慢、不稳,先后就诊于多家医院未明确诊治,后就诊于我院行头部CT,眼科检查双眼K-F环(+),血清铜蓝蛋白降低且24 h尿铜含量升高,诊断为“肝豆状核变性”,给予驱铜、护肝、营养神经等治疗,期间因脾大、脾亢行“脾切除术”治疗。2008年初,因未遵医嘱服用药物,逐渐出现脾气暴躁、易怒,动辄打人毁物,加用“氯丙嗪”控制精神症状。2011年后相继自行停用氯丙嗪、驱铜等药物,病情尚稳定。9 m前,患者言语不清伴行走不稳症状再次加重,且出现肢体扭转伴疼痛。发病以来,饮食、睡眠尚可,二便如常。父母体键,否认近亲结婚史,患者一哥哥产后全身皮肤黝黑,出生3 d死亡(未查病因)。家族成员中否认类似病史。内科查体:全身皮肤黏膜及双巩膜无黄染,腹平软,腹中部可见一长约20 cm陈旧性手术疤痕,肝肋下未触及,脾已切除,移动性浊音(-),双下肢无水肿。神经系统查体:神志清楚,说话缓慢而含糊,发音单调,发音费力,仅可简单应答。双眼球活动自如,无眼震。伸舌不能,双侧软腭抬举减弱,软腭反射迟钝。左上肢、双下肢屈曲扭转畸形。四肢肌力5级。四肢肌张力呈铅管样增高。双侧病理反射未引出。余查体不合作。
辅助检查:肝功:门冬氨酸氨基转移酶50.9 U/L;肾功:肌酐114.5 μmol/L;血脂:甘油三脂1.81 mmol/L,高密度脂蛋白0.94 mmol/L,低密度脂蛋白3.75 mmol/L;铜生化:铜蓝蛋白33.7 mg/L,铜1.3 μmol/L,铜氧化酶0.021OD。24 h尿铜175.1 μg。头部CT(见图1):双侧苍白球区、桥脑低密度影。腹部彩超:(1)肝豆肝病样改变(结节型);(2)胆囊炎;(3)右肾结石。腹腔血管彩超:门静脉主干内未见明显血栓形成。眼部检查:K-F环(+)。基因检测:ATP7B基因p.R778L杂合突变,一个意义未明的内含子区剪切突变(见图2)。肌电图:可见左肱二头肌、尺侧腕屈肌、指短伸肌及右拇长伸肌成组中量MUP阵发性发放。UWDRS评分量表:神经功能评分112分,精神症状评分22分。诊断:肝豆状核变性(混合型),ATP7B基因p.R778L杂合突变。患者在外院进行驱铜治疗,1 m后症状好转,但未能按疗程完成治疗,1 y后电话随访,患者症状再次加重,建议其系统治疗。
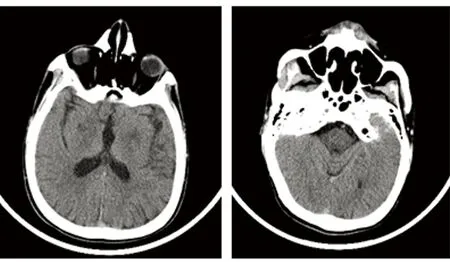
图1 双侧苍白球区、桥脑低密度影

图2 ATP7B基因p.R778L杂合突变
2 讨 论
英国神经病学家Samuel Wilson[2]于1912年首次发现肝豆状核变性,是位于13号染色体的ATP7B基因突变,使其编码的P型ATP酶功能减弱甚至消失,致胆道排铜障碍及血清铜蓝蛋白合成减少,大量铜沉积在不同的组织中,典型症状为进行性加重的神经系统表现、肝脏疾病和眼部改变。
2008年《肝豆状核变性诊断与治疗指南》[3]将肝豆状核变性分为4型:脑型、肝型、其他类型及混合型。Wilson病主要累及中枢神经系统称为脑型Wilson病,多数表现为运动功能障碍,比如原因不明的锥体外系症状,特别是手部远端不对称性震颤或头部、躯干震颤、肌张力增高、共济失调等,也可出现流涎、构音障碍、吞咽困难,严重者会出现性格改变、精神分裂等精神症状。其中将脑型WD又划分为典型WD型、扭转痉挛型、精神障碍型及假性硬化型。WD发病年龄通常>15岁,也有报道为7~9岁[4],本例患者发病时16岁,以构音障碍、共济失调为首发症状,逐渐出现精神症状,精神症状是非特异性的,可以表现为抑郁、躁狂,或仅仅为情绪不稳定。故当青少年患者出现不能解释的认知障碍、精神障碍或者运动障碍需排除此病。
肝脏发生病理改变为主的称为肝型Wilson病,表现为肝炎、肝硬化及肝衰竭等临床症状。K-F环[5],即Wilson病患者角膜处黄棕色或黄绿色色素环,是由于铜颗粒沉积于角膜后弹力层形成的,几乎见于所有出现神经病变的患者中,为该病最常见的眼部体征,但K-F环阴性也不能排除WD的可能。此外,也可造成肾脏损伤、骨关节肌肉损害或溶血性贫血,属于其他类型Wilson病。混合型WD是以上各型的组合。
本例患者为青年男性,具有肝豆状核变性经典的“三联征”,累及中枢神经系统、肝脏及眼部,且病情持续进展。特别之处在于,本例患者以神经损伤为突出表现(脑型),同时肝实质改变(肝型),肌酐升高提示肾脏损害(其他类型),为临床比较少见的混合型Wilson病。目前国内外有关混合型WD报道较少,吕佳等[6]2019年报道了以神经障碍为突出症状的肝豆状核变性1例,此患者以神经损伤为突出表现,同时存在肝硬化,尿常规见隐血、肌酸激酶升高提示肾脏、骨骼肌损害。神经系统表现具有较大的临床异质性,最常见的神经系统异常包括构音障碍、肌张力障碍、震颤和帕金森病,疾病的非典型表现增加了诊断的复杂性。患者肝实质发生了肝豆肝病样改变(结节型),肝功化验中门冬氨酸氨基转移酶略有异常,考虑与患者长期驱铜、服用护肝片等对症治疗有关;患者肌酐高于正常,说明铜对肾脏也造成了一定程度的损伤。铜蓝蛋白是一种主要由肝脏表达合成的含铜的α2球蛋白,运输血液中90% 以上的铜至各个器官,参与人体内铜、铁等微量元素的平衡,患者的铜蓝蛋白及血清铜、尿酮均发生相应改变。
对该患者进行了基因检测,第8号外显子测序显示,ATP7B基因编码区检出c.2333G>T杂合突变,是我国肝豆状核变性患者常见基因突变位点。HLD患者的基因水平确诊需满足ATP7B基因纯合致病突变或复合杂合致病突变,单一杂合突变不能确诊[7]。本患者ATP7B基因不符合确诊条件,其患者父母也未能行基因检测,但全球发现WD患者ATP7B基因突变类型700余种,少数属于内含子突变,尤其是剪切突变,故只检测突变热点用于诊断和筛查,容易漏诊[8],而且WD诊断是从基因到临床的综合统一,不是靠单一指标。本患者基因检测出4号内含子区c.1708-1G>C剪切突变,但意义未明,提示我们需要对更多的家系进行基因研究,以便更全面地了解WD患病情况。
本研究报道的病例应该引起对HLD的不同分型及起病形式多样更充分的认识,临床医生应仔细询问病史,同时检查血清铜蓝蛋白、血铜、24 h尿铜、角膜K-F环及ATP7B基因等多项指标,减少误诊、漏诊,Wilson病属于目前少数几种可控的神经遗传病之一,使患者得到早诊断、早治疗,最终改善疾病预后。
