山东省9 147例新生儿耳聋基因和听力联合筛查结果分析
2020-09-17孙毅刘雅琳刘晓莉丁美玲张斌
孙毅 刘雅琳 刘晓莉 丁美玲 张斌
耳聋是影响人类健康和造成人类残疾的常见原因,研究发现65%的耳聋是由遗传引起的[1];其可以由单一基因突变或多基因突变引起,也可以由环境因素或基因和环境因素共同导致。随着新生儿听力筛查工作的开展,发现常规听力筛查存在一定的局限性,不能及时发现迟发性耳聋。北京市新生儿遗传性耳聋基因筛查研究发现25%的遗传性耳聋被单纯的听力学筛查遗漏;耳聋基因筛查联合听力筛查有助于预知迟发性耳聋和药物性耳聋,同时做到有目的性的随访和临床干预[2~5]。本研究拟通过分析近年来山东地区新生儿听力及耳聋基因联合筛查结果,探讨该地区新生儿耳聋基因联合听力筛查的临床意义,为该方法的推广应用提供参考。
1 资料与方法
1.1研究对象 以2017年6月至2018年11月在山东省内各地分娩的新生儿9 147例为研究对象(不含重症监护室新生儿),筛查前明确告知听力筛查和耳聋基因筛查相关知识,监护人或家属签订知情同意书。
1.2新生儿听力筛查及诊断方法
1.2.1听力筛查和诊断流程 新生儿在出生48小时后,在自然睡眠状态下,于环境噪声小于40 dB A的房间使用畸变产物耳声发射(DPOAE)进行听力初筛,初筛未通过者要求在30~42天内使用自动听性脑干反应(AABR)进行听力复筛。复筛未通过者将检测结果告知家长,并要求3月龄时应用听性脑干反应(ABR)进行听力诊断。
1.2.2听力筛查方法 使用德国MAICO公司生产的Ero scan型耳声发射仪进行初筛,①DPOAE检测,测试时间5~7秒,刺激强度40~70 dB SPL,测试频率2~5 kHz,设备自动显示“通过”或“未通过”“转诊”。②AABR检测:采用德国MAICO的MB11筛查AABR模块进行复筛,刺激强度35 dB nHL,测试频率0.75~5 kHz,刺激信号为CE-Chirp声,仪器自动显示 “通过”或“未通过”“转诊”。
1.2.3听力诊断方法 复筛未通过者清除外耳道耵聍后,进行听力学诊断。ABR检测:采用德国MAICO生产的MB11型检测仪标准ABR模块。以反应阈<30 dB nHL为正常,31~50 dB nHL为轻度听力损失,51~70 dB nHL为中度听力损失,71~90 dB nHL为重度听力损失,>91 dB nHL为极重度听力损失。
1.3耳聋基因检测方法 新生儿出生3天后,由各助产机构用采血卡滤纸采集新生儿足跟末梢血2个血斑,每个血斑直径不小于8 mm,室温下自然晾干后装入带速封条的塑料袋内,每周派专人送至山东省康复研究中心耳聋基因筛查实验室对标本进行检测,共检测4个常见的耳聋基因(GJB2、GJB3、SLC26A4和mtDNA12SrRNA),包括15个耳聋突变位点,GJB2(35delG、 176del16、235delC、299_300delAT)、SLC26A4 (1174A>T、1226 G>A、1229 C>T、1975 G >C、2027T>A、 IVS15+5G>A、IVS7-2A>G、2168A>G)、mtDNA12SrRNA(1494C>T、1555A>G)、GJB3(538C>T),并在30天内将检测结果反馈至新生儿出生分娩机构。
检测流程:采用天根干血斑核酸提取试剂盒(DP334-03)对干血斑进行基因组DNA提取,使用Nano DRrop2000对提取的DNA进行浓度检测,浓度合格后使用PCR扩增仪对目的基因片段进行扩增(杭州博日科技有限公司TC-96/G/H(b)),后使用晶芯BioMixer TM 芯片杂交仪、晶芯SlideWasher TM洗干仪和LuxScan TM 10K-A微阵列芯片扫描仪和遗传性耳聋基因芯片判别系统(博奥晶典生物技术有限公司)对检测结果进行判读。每批样本包含阴性、阳性对照组,以验证芯片检测结果的可靠性,同时随机抽取5%的样本以及检测结果为阳性的样本进行DNA测序,进一步验证芯片检测结果的准确性。
2 结果
2.1听力筛查及诊断结果 9 147例新生儿中,初筛未通过172例(1.88%,172/9 147),复筛未通过者94例(54.65%,94/172),经听力学诊断确诊听力损失患儿17例(表1),占初筛未通过的9.88%(17/172);其中,双耳听力损失10例,包括极重度2例,重度2例,中度1例,轻度4例,左耳轻度、右耳中度1例。单耳听力损失极重度1例,中度2例,轻度1例。先天性小耳畸形2例,面部发育畸形、眼距异常伴双耳极重度听力损失1例。
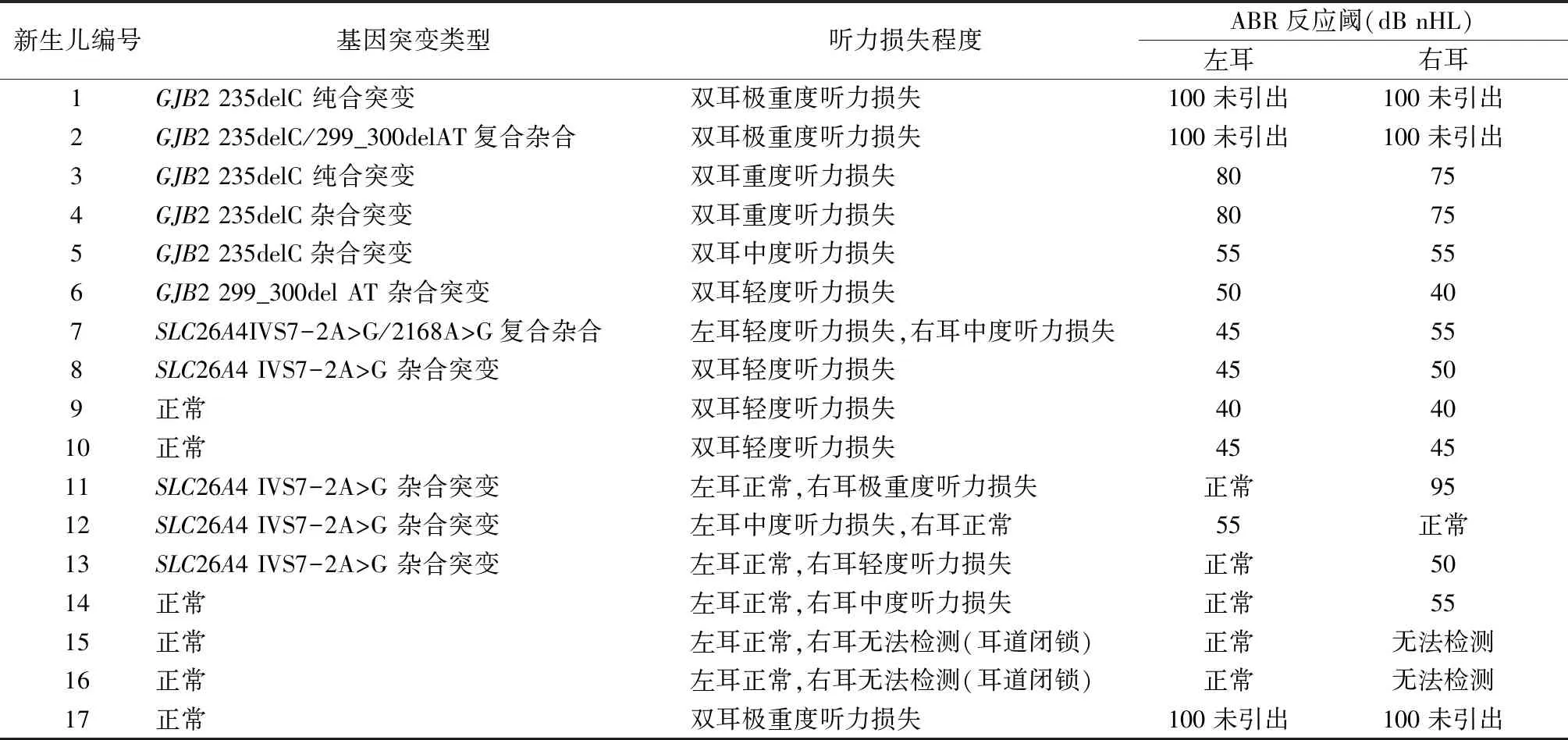
表1 17例诊断听力损失新生儿基因筛查突变类型、听力损失程度及ABR反应阈
2.2基因筛查结果 9 147例新生儿全部进行十五个位点的耳聋基因筛查,发现耳聋基因突变461例(表2)。致聋基因携带率5.04%(461/9 147)。筛查出GJB2基因杂合突变224例,携带率为2.45%,GJB3基因杂合突变27例,携带率0.30%;SLC26A4基因杂合突变186例,携带率2.03%;mtDNA12SrRNA均质和异质突变19例,携带率0.21%。GJB2纯合突变2例,复合杂合突变1例,SLC26A4基因复合杂合突变1例。双基因杂合突变5例。对耳聋基因检测结果阳性患儿家长进行电话回访,在461例阳性患儿中有103位家长未取得联系,剩余358例耳聋基因检测阳性患儿中19例未通过听力初筛,进行了听力学诊断,结果显示有11例耳聋基因检测阳性患儿听力诊断异常。
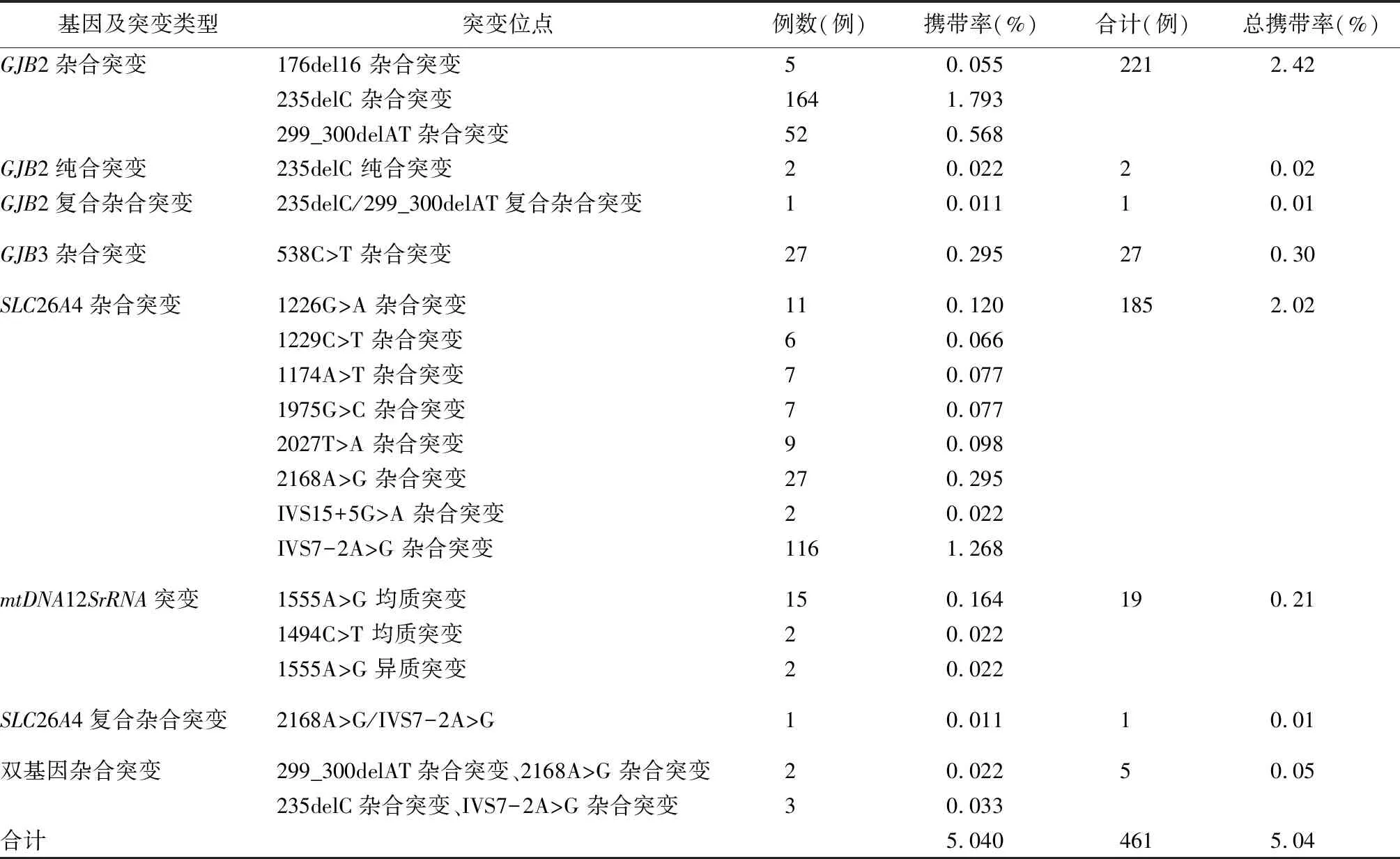
表2 山东省9 147例新生儿耳聋基因筛查各基因突变类型、位点及携带率
2.3确诊听力损失患儿的基因筛查结果 在17例听力损失患儿中耳聋基因检测阴性6例,阳性11例,11例耳聋基因筛查阳性者中,携带GJB2基因突变者,包括:2例235delC纯合突变,1例235delC/299_300delAT复合杂合突变,2例235delC杂合突变,1例299_300delAT杂合突变;携带SLC26A4基因突变者,包括1例IVS7-2A>G/2168A>G复合杂合突变,4例IVS7-2A>G杂合突变(表1)。在6例基因检测阴性患儿中,1例面部发育异常提示综合征型耳聋。根据家长要求,对1例GJB2基因299_300delAT杂合突变且听力诊断为双耳轻度听力损失患者进行基因组测序检查发现, 其携带257C>G突变,为复合杂合突变,明确了遗传学病因。
3 讨论
研究发现65%的耳聋是由遗传引起的[1]。新生儿听力筛查经过多年发展,已经比较成熟,但是,新生儿听力筛查有一定局限性,迟发性耳聋及药物性耳聋患者均可通过新生儿听力筛查。70%的遗传性耳聋是以耳聋为唯一症状而不伴有其他临床表现[6],耳聋基因诊断则有助于判别是否为遗传导致的耳聋[7]。
从文中结果看,山东省9 147例新生儿耳聋基因携带率为5.04%,以GJB2和SLC26A4基因突变为主,携带率分别为2.45%和2.03%,GJB3和线粒体DNA12SrRNA基因突变携带率分别为0.30%和0.21%,其中GJB2、GJB3和线粒体DNA12SrRNA基因突变携带率与王秋菊等[2]的研究结果基本一致,只有SLC26A4结果略有不同。总结可能有两方面原因,一是本研究使用芯片在SLC26A4位点上比之前的研究多了6个位点,有利于发现更多突变个体;二是我国地域广阔,各个地区之间耳聋基因突变的分布频率可能存在一定差异。
在我国人群中,GJB2基因是引起遗传性耳聋最常见的耳聋基因[1]。GJB2基因编码的连接蛋白,主要参与细胞间信号介导和离子的传递[8],其表达或者功能的异常会影响细胞间隙的连接功能,影响内耳钾离子的正常回收而致聋;其遗传方式表现为常染色体隐性遗传,235delC突变在听力正常人群中携带率为1%~2%[9]。本组新生儿GJB2基因突变发生率最高,携带率为2.45%,其中有164例为235delC杂合突变,携带率达1.79%,此结果与文献报道相一致。本次筛查中发现2例GJB2纯合突变和1例GJB2复合杂合突变的新生儿,这3例听力初筛及复筛均显示为“未通过”或“转诊”,听力诊断2例为极重度听力损失,1例为重度听力损失。另外,还发现1例299_300delAT杂合突变者,双耳为轻度听力损失,对该例新生儿进行GJB2全序列测序,发现同时携带257C>G突变,是复合杂合突变,明确了遗传学病因[10],故建议其尽早进行听力干预并长期随诊。研究报道,有50%以上的极重度耳聋患者在3月龄时具有正常的听觉行为,至少20%在6个月时仍具有听觉行为[11]。因此,上述病例需进行随访,追踪其听力变化情况,以免延误治疗和干预。
SLC26A4基因又称PDS基因,位于人类染色体7q31,含21个外显子,是大前庭水管综合征的主要致病基因,其编码的Pendred蛋白为离子转运体,主要由疏水性氨基酸组成,其功能主要参与碘/氯离子等阴离子的转运[12]。SLC26A4基因是仅次于GJB2突变而引起感音神经性聋的遗传学病因,遗传方式为常染色体隐性遗传[13]。本组9 147例新生儿中SLC26A4基因携带者186例,其中发现1例IVS7-2A>G/2168A>G复合杂合突变患者,双耳均通过听力筛查,召回其父母采血行耳聋基因检测,结果显示父母分别为IVS7-2A>G杂合突变携带者和2168A>G杂合突变携带者;该患儿于出生后6月龄时行听力诊断,CT结果示其右耳前庭水管扩张,左耳正常,听力诊断结果示左耳轻度听力损失,右耳中度听力损失,建议患儿及时佩戴助听器,该夫妇再次生育时应进行产前诊断。研究发现单纯IVS7-2A>G杂合突变也有导致中度语后聋和重度语前聋的报道,很可能是SLC26A4基因上的第二个突变位点没有被发现[14]。本研究中发现4例SLC26A4杂合突变个体同样存在双侧或单侧的听力损失,后期通过影像学诊断发现全部为大前庭水管综合征,将在后续的研究中对这些患儿进行SLC26A4基因全基因测序分析,找到第二个可能的突变位点。
SLC26A4基因又称为“一巴掌打聋基因”。在患儿成长过程中,头部运动、外力作用、巨大的声响及感冒、擤鼻涕等因素都可能导致听力障碍的发生。本次筛查中检出的SLC26A4基因突变186例,携带率为2.03%,仅次于GJB2的突变携带率;其中SLC26A4基因IVS7-2A>G突变116人,占SLC26A4基因突变62.7%。除上述1例IVS7-2A>G/2168A>G复合杂合突变患者及4例携带SLC26A4致聋基因听力筛查未通过者外,剩余181例SLC26A4基因携带者均建议其进行听力诊断检查或该基因全系列分析。通过基因检测,对于已经确诊的患儿不仅可以早期明确病因诊断,还可以针对性地给予听力保健指导。同时有利于提醒听力检测通过而基因检测未通过的新生儿家长关注小儿听力变化,如果发现听力损失应及早保护患儿的残余听力并给予早期干预。
mtDNA12SrRNA突变为母系遗传,mtDNA12SrRNA基因1494C>T、1555A>G与药物性聋有关[15],1555A>G、1494C>T两个突变可以导致12SrRNA的构象改变,形成与氨基糖苷类抗生素作用的结合位点,进而导致听毛细胞逐渐凋亡引起永久性不可逆的耳聋。本研究在山东地区9 147例新生儿标本中发现mtDNA12SrRNA突变患者19例,均通过听力诊断,在检出结果后及时对该基因突变患者及其母系家庭成员的临床用药进行宣教,要求在后续的生活中禁止使用耳毒性药物以防“一针致聋”。通过本次筛查可避免19位听障患儿的产生,同时对家系成员进行用药指导,避免后代产生听力残疾患者。
GJB3基因最初被发现在中国患者中导致常染色体显性遗传非综合征型聋。2013年我国学者首次报道了新生儿中该基因的筛查结果[16],目前认为该基因突变携带者可能为迟发性耳聋患者,虽然在新生儿期无耳聋症状,但应密切关注其听力状况。本研究发现27例为GJB3基因538C>T杂合突变,其听力筛查结果正常,建议定期随访。
本研究结果表明,新生儿听力筛查联合耳聋基因筛查是大势所趋,通过基因筛查可以早期发现部分听力筛查不能发现的迟发性耳聋个体和药物性耳聋个体及家系,扩大干预范围,同时能够依据发现的遗传信息进行婚育指导,使防聋时间关口前移。目前,对新生儿耳聋基因筛查和听力筛查的宣传教育还是集中于医疗机构[17],经由医务人员对家长进行宣教,尚不够普及,效果受限。因此,残疾人服务机构更应该加入到残疾预防的行列中,积极通过多渠道提高本地区家长对遗传性耳聋的认识,科学的理解遗传性聋的发病原因,避免因为认识不足造成的延误诊断,科学的面对听力残疾预防及残疾康复。
