胶基型嚼烟中19种多环芳烃的气相色谱—串联质谱技术法测定
2020-07-03蔡洁云王惠平顾健龙孙浩巍
蔡洁云 王惠平 刘 巍 顾健龙孙浩巍 张 轲 龙 杰 李 超
(1. 云南省烟草质量监督检测站,云南 昆明 650104;2. 云南中烟工业有限责任公司技术中心,云南 昆明 650024)
胶基型嚼烟是一种通过咀嚼提供烟碱的新型烟草制品产品,是一种重要的无烟气烟草制品[1],主要含有4类成分:可咀嚼的天然树胶和热塑性树脂等食用胶基、烟草(烟草提取物)、烟碱,以及香精香料、软化剂、甜味剂等食品添加剂[2]。目前,对无烟气烟草制品成分及其含量的检测尚未形成相关标准,研究成果也有限。对无烟气烟草制品成分的研究[3-5]主要侧重于烟碱含量及其释放行为,也有一些成果对挥发性香味物质[6]、醛类物质[7]、TSNAs[8-9]、多环芳烃类物质[10]、多元醇类保润剂[11]、重金属元素[12]的检测进行了报道。
多环芳烃(Polycyclic Aromatic Hydrocarbons,PAHs)是一类分子中含有两个或两个以上苯环以线状、交状或簇状排列形成的稠环形芳香化合物,是难降解、易挥发的碳氢化合物,多具致癌性[13-14]。国际癌症研究机构(IARC)[15-17]对卷烟中多环芳烃的动物致癌试验表明,苯并[a]芘、苯并[k]荧蒽、苯并[g,h,i]芘和茚并[1,2,3-cd]芘等多环芳烃具有强致癌性;其中,苯并[a]芘致癌性最强,属于一类致癌物质,其在较低浓度下就有强致癌作用,可能诱发食管癌、胃癌,或经空气吸入诱发肺癌。近年的研究表明,无烟气烟草制品的细胞毒性[18]和致癌性[19]小于传统卷烟。最近的研究[20]证实,无烟气烟草制品中含有多种多环芳烃。对无烟气烟草制品中多环芳烃含量检测鲜有研究,对胶基型嚼烟中多环芳烃类物质的含量检测尚未见文献报道。多环芳烃检测前处理过程通常需要固相萃取对样品进行净化,操作繁琐且耗时长。
试验拟采用串联质谱检测器,以目标物的质荷比为导向,建立一种简便高效的测定胶基型嚼烟中19种多环芳烃(包括两环芳烃2个、三环芳烃5个、四环芳烃4个、五环芳烃6个、六环芳烃2个)含量的方法,以期为胶基型嚼烟质量控制提供参考。
1 材料与方法
1.1 材料与试剂
胶基型嚼烟样品:6种市售进口胶基型嚼烟样品,试验前样品密封储存于冰箱中(-18 ℃);
萘(NAP)、1-甲基萘(1-Me NAP)、苊烯(ANY)、苊(ANA)、芴(FLU)、菲(PHE)、蒽(ANT)、荧蒽(FLT)、芘(PYR)、苯并[a]蒽(BaA)、屈(CHR)、苯并[b]荧蒽(BbF)、苯并[k]荧蒽(BkF)、苯并[e]芘(BeP)、苯并[a]芘(BaP)、苝(PER)、茚并[1,2,3-cd]芘(IPY)、二苯并[a,h]蒽(DBA)、苯并[g,h,i]芘(BPE)、氘代萘(NAP-d8)、氘代蒽(ANT-d8)、氘代苯并[a]芘(BaP-d8)标准品:纯度≥99%,美国Sigma-Aloaich公司;
环己烷:色谱纯,美国Fisher Scientific公司。
1.2 主要仪器设备
电子天平:AG204型,瑞士梅特勒—托利多公司;
振荡器:MAXQ 2000型,赛默飞世尔科技(中国)有限公司;
气相色谱—三重串联四极杆质谱联用仪(GC-MS/MS):7890A/7000B型,安捷伦科技(中国)有限公司。
1.3 样品前处理
试验前,将待分析样品在室温下解冻2 h。解冻后,将样品切块、研磨粉碎后,过筛(颗粒直径<4 mm),准确称取1.00 g 样品置于50 mL 锥形瓶中,加入含有氘代萘(320 ng/mL)、氘代蒽(320 ng/mL)和氘代苯并[a]芘(320 ng/mL)的环己烷溶液250 μL作为内标物质,再加入10 mL环己烷作为萃取剂;将锥形瓶在室温下振荡萃取40 min后,静置5 min,将萃取液通过0.22 μm尼龙66滤膜,收集样品于色谱瓶中,待GC-MS/MS分析。
1.4 GC-MS/MS检测参数
色谱进样口温度:260 ℃;进样量:1 μL;进样方式:不分流进样;程序升温:初始温度40 ℃,保持7 min;以20 ℃/min升至100 ℃;以4 ℃/min升至290 ℃并保持20 min;传输线温度:280 ℃;质谱电离方式:EI;离子源温度:280 ℃;质谱扫描方式:动态多反应监测(dMRM)模式;碰撞气:氮气,流速1.5 mL/min;载气:氦气,流速2.25 mL/min。
以最高浓度的工作溶液为分析对象,通过全扫描模式(MS1 scan)选择各目标物及内标物质合适的母离子;在此基础上,在5~60 eV的条件下,以5 eV为检测间隔,采用产物离子扫描模式(product ionscan)进行检测,优化碰撞能量,优选产物离子[21]。待测化合物及其内标的质谱分析参数见表1。
1.5 结果的计算与表述
将标准系列溶液中待测物的含量与内标量之比对色谱图中待测物峰面积与内标面积的比值作图,得到工作曲线的a值和b值。样品中的待测物含量按式(1)计算,取两个平行样品的算术平均值作为样品的测试结果。含量以每克胶基型嚼烟中多环芳烃的质量表示,单位为ng/g,结果精确到0.01 ng/g。
(1)
式中:
mi——胶基型嚼烟中多环芳烃物质i的含量,ng/g;
Ai——萃取液中多环芳烃物质i的峰面积;
a、b——由线性回归方程求出;
Ms——样品溶液中的内标含量,ng;
As——内标峰面积;
V——萃取液的体积,mL;
m——进行试验分析的胶基型嚼烟,g。
取两次平行测定结果的算术平均值作为样品的最终测试结果,精确至0.01 ng/g。
2 结果与讨论
2.1 GC-MS/MS检测条件的选择
无烟气烟草制品中PAHs的含量范围较广,不同牌号产品中的PAHs含量差异很大,基质复杂。为确保方法的稳定性和重复性,对于19种PAHs都通过选定2个离子对(1个定性离子对和1个定量离子对)和相应的氘代内标来确保方法的准确性,并且每个PAHs都选用相应的氘代内标对其进行准确定量,使得定量内标在保留时间和峰形上都能与定量离子保持较好的一致性(见表1)。
由于同环数的PAHs化学结构及性质类似,三环的菲和蒽,四环的荧蒽和芘,四环的苯并[a]蒽和屈,五环的苯并[b]荧蒽、苯并[k]荧蒽、苯并[e]芘、苯并[a]芘和苝,六环的茚并[1,2,3-cd]芘和苯并[g,h,i]芘,上述5组分别为同分异构体,需要选择适宜的色谱条件,使每组同分异构体中各化合物的色谱峰完全分离,才能准确定量分析。最终优化的色谱条件如1.3所述,在上述条件下,所有待测化合物和内标物质都能很好的质子化,色谱峰型均较好(见图1)。
2.2 萃取时间的选择
PAHs多易溶于环己烷,因此环己烷作为萃取试剂。

表1 化合物信息及质谱分析参数
为选择合适的萃取时间,按1.3所述前处理步骤,准确称取5份相同的样品,加入内标溶液和10 mL环己烷后,进行常温振荡萃取,萃取时间分别为20,30,40,50,60 min。其检测结果如图2所示。
由图2可以看出,萃取时间为20,30 min时,待测物并未完全溶解至萃取液中;当萃取时间>40 min时,大部分待测物在萃取液中的含量趋于稳定,小部分待测物在萃取液中的含量呈峰值状态。因此,较优的萃取时间为40 min。
2.3 方法学评价
2.3.1 线性关系与检出限、定量限 建立标准工作曲线的一系列标准工作溶液中,含萘、1-甲基萘、苊烯、苊、芴、菲、蒽、荧蒽、芘、苯并[a]蒽、屈、苯并[b]荧蒽、苯并[k]荧蒽、苯并[e]芘、苯并[a]芘、苝、茚并[1,2,3-cd]芘、二苯并[a,h]蒽、苯并[g,h,i]芘的标准物及其内标氘代萘、氘代蒽和氘代苯并[a]芘,溶剂为环己烷。根据试验检测所得胶基型嚼烟中多环芳烃的含量范围,确定标准工作溶液浓度依次为0.5,1.0,2.0,5.0,10.0,20.0,50.0,200.0 ng/mL(标准工作溶液各分析物的浓度相同);标准工作溶液中内标浓度均为8 ng/mL。
将所配制的PAHs标准溶液由低到高浓度进样分析,以分析物与内标物的峰面积比(Y)对相应的分析物浓度(X,ng/mL)进行线性回归分析,得到各分析物的工作曲线回归方程和相关系数。PAHs的线性范围为0.5~200.0 ng/mL,线性相关系数为0.998 7~0.999 9,表明在测定的浓度范围内具有良好的线性关系。取最低浓度标准溶液(0.5 ng/mL),平行测定10次,计算测定值的标准偏差(SD),以3SD计算方法的检出限[22],分别为0.04~0.14 ng/mL;以10SD计算方法的定量限,分别为0.14~0.46 ng/mL,结果见表2。
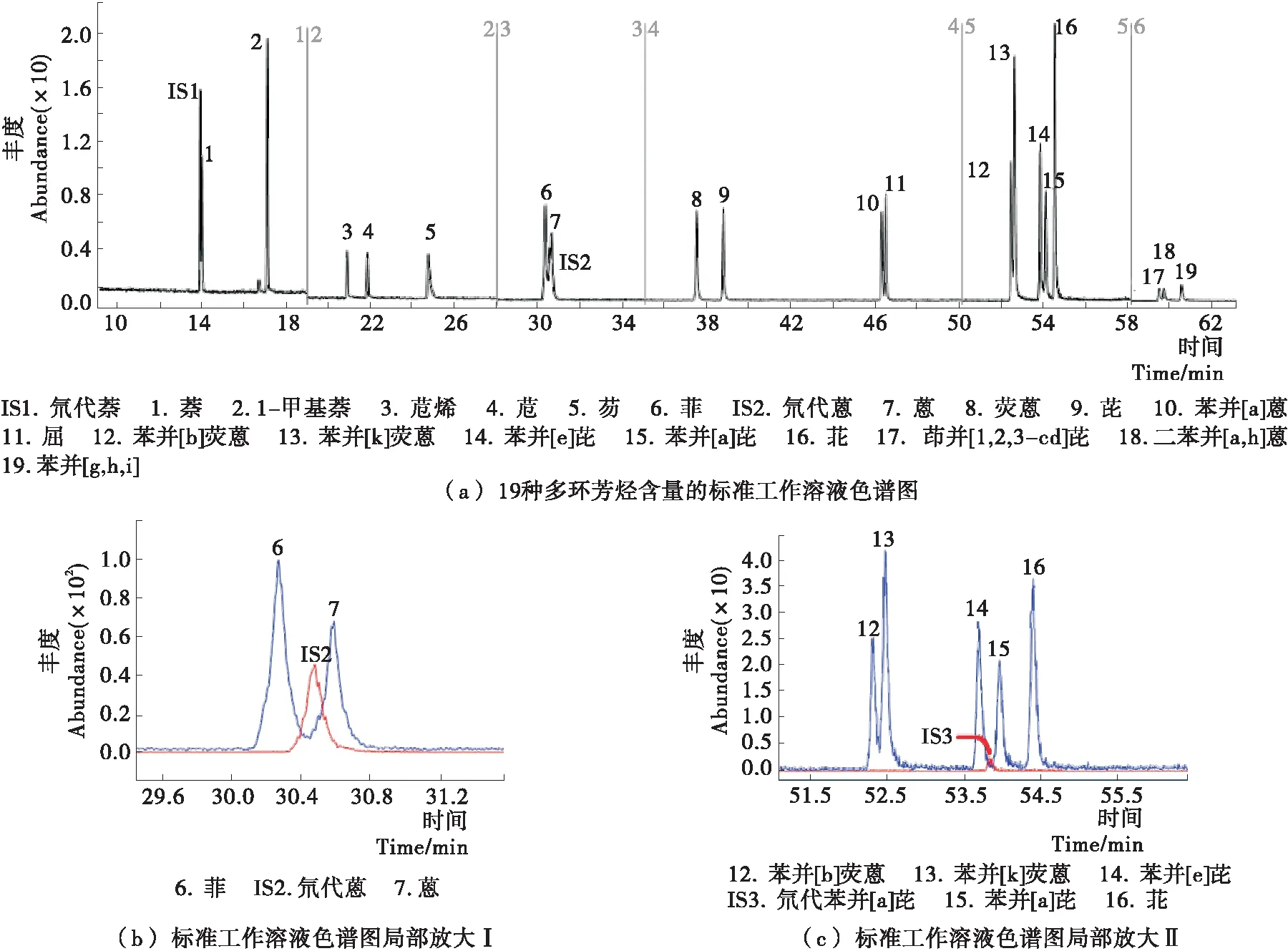
图1 GC-MS/MS法测定胶基型嚼烟中19种多环芳烃含量的标准工作溶液色谱图
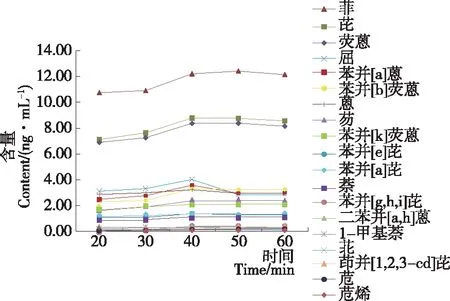
图2 萃取时间对胶基型嚼烟样品检测结果的影响
Figure 2 Effects of shake duration on the detection results of gum-based chewing tobacco
2.3.2 准确性和重现性 经优化条件,测定低、中、高3个添加水平的胶基型嚼烟样品中19种多环芳烃,重复进行6次加标回收率试验,计算各物质的回收率,结果见表3。结果表明,在3种添加水平下,19种多环芳烃的加标回收率分别为92.26%~109.10%,92.85%~109.46%,97.42%~106.17%,测定结果的相对标准偏差为0.77%~4.70%,表明该方法准确性较高。
采用加标样品(中等浓度加标)进行稳定性的精密度考察,对各样品分别进行6次平行测定,根据结果分别计算各多环芳烃的相对标准偏差(RSD)。结果显示,日内和日间RSD范围分别为0.77%~3.85%,1.57%~4.86%,均<5%,说明所建方法重现性较好。
2.4 胶基型嚼烟样品的PAHs含量测定
采用优化分析方法,测定了6种胶基型嚼烟样品中各种PAHs含量。检测结果表明,样品中多环芳烃均未检出(n=2)。
试验结果表明,从多环芳烃类物质传递的角度而言,由于生产工艺和原料的影响,胶基型嚼烟的溶出物中一般不含有多环芳烃类物质,属于较为安全的无烟气烟草制品。
3 结论
试验建立了一种应用气相色谱—串联质谱技术(GC-MS/MS)技术测定胶基型嚼烟中多环芳烃含量的方法,以内标法定量。该方法具有样品前处理简单快捷、试剂用量少、灵敏度高等优点,且可以同时检测胶基型嚼烟中19种多环芳烃物质,能满足胶基型嚼烟中多环芳烃简单、快速、准确检测的要求。该方法还可推广至含烟、鼻烟等其他类型的无烟气烟草制品的检测中。

表2 19种PAHs的工作曲线回归方程、R2、检出限和定量限

表3 GC-MS/MS法测定胶基型嚼烟中多环芳烃的加标回收率和精密度†
† 低添加水平1 ng/mL,中添加水平20 ng/mL,高添加水平200 ng/mL。
