基于皮克林乳液聚合四环素磁性分子印迹—生物炭微球的研制
2020-07-03马珍珍何金兴赵晓磊
马珍珍 何金兴 赵 涛 赵晓磊
(齐鲁工业大学〔山东省科学院〕食品科学与工程学院,山东 济南 250353)
四环素类抗生素(Tetracyclines,TCs)是一类临床应用较早的广谱抗生素。由于TCs成本低且较易获得,被广泛应用于防治动物疾病和促进动物生长[1],不合理的滥用导致TCs在动物源性食品中残留严重,对人体健康和环境具有潜在危害,如过敏反应[2]、抗药性基因的出现和传播[3],增加患皮肤癌的风险[4]。GB 31650—2019中明确规定在禽蛋中TCs最大残留限量为400 μg/kg,动物肌肉组织中为200 μg/kg,肝脏中为600 μg/kg,牛奶中为100 μg/kg。目前,针对TCs的定量分析方法包括色谱法[5]、荧光分析法[6]、免疫分析法[7]、微生物抑制法[8]和传感器分析技术[9]等。其中,色谱法是最常用的检测技术,该方法的测量准确度高、重现性好,但因食品基质中干扰成分多且分析物丰度低,样品前处理过程对检测结果的准确性和灵敏度有很大影响。
生物炭作为一种新兴吸附材料,已被证实可用于去除环境中氯霉素和四环素(TC)污染[10-11]。生物炭来源广泛、价格低廉、制备简单,具备丰富的多孔结构且易于修饰[12],在食品样品前处理领域具有广阔的应用前景。但实际应用中,生物炭缺乏特异性的劣势给食品中痕量目标物的识别带来了难度。分子印迹技术是模拟抗体—抗原的识别过程,制备高特异性的聚合物材料的方法,常规的分子印迹材料的制备方法包括本体聚合、原位聚合和表面印迹等[13]。皮克林乳液是以固定颗粒作为稳定剂形成两相或多相体系,由于固体颗粒在水相和在油相的亲润性不同,能够稳定吸附在两相界面上阻止液滴的碰撞和合并[14-15]。利用皮克林乳液聚合法制备生物炭印迹聚合物,通过两相界面上印迹方法,可以改善生物炭在常规溶剂中分散性差并解决常规方法合成过程中生物炭可能团聚导致印迹位点分布不均的问题。
试验拟以生物炭作为皮克林乳液稳定剂,制备新型水包油型皮克林乳液。以TC为模板分子、甲基丙烯酸为功能单体、二乙烯基苯为交联剂、偶氮二异丁腈为引发剂,四氧化三铁(Fe3O4)与生物炭作为共稳定剂,制备磁响应四环素印迹生物炭微球(MMIPMs)。并以此聚合物为磁性固相萃取材料,与高效液相色谱仪(HPLC)离线联用,建立适用于动物源性食品中TCs痕量残留检测的方法。旨在为生物炭在食品中痕量污染物检测方面的应用提供一种新思路和新方法。
1 材料与方法
1.1 材料与仪器
1.1.1 材料与试剂
TC:98%,梯希爱(上海)化成工业发展有限公司;
盐酸土霉素(OTC)、盐酸强力霉素(DC):98%,上海源叶生物科技有限公司;
磺胺二甲基嘧啶(SMZ,99%)、甲基丙烯酸:西格玛奥德里奇(上海)贸易有限公司;
速灭威(TMC,99.2%)、偶氮二异丁腈:天津市科密欧化学试剂有限公司;
二乙烯基苯:80%,上海易恩化学技术有限公司;
生物炭:400~450 ℃热解的稻壳生物炭,沈阳卡力玛生物炭科技开发有限公司;
鱼和鸡肉样品:市售,用均质机均质后置于-20 ℃冰箱保存备用。
1.1.2 主要仪器设备
HPLC:1260 Infinity II型,美国Agilent Technologies公司;
扫描电子显微镜:EM-30plus型,韩国COXEM公司;
X射线衍射仪:D8-Advance型,德国Bruker公司;
热重分析仪:TGA-1型,瑞士Mettler公司;
傅里叶红外光谱仪:Nicolet 6700型,美国Thermo Fisher Scientific公司;
紫外可见分光光度计:TU1901型,北京普析通用仪器有限责任公司。
1.2 方法
1.2.1 二乙烯基苯纯化 取200 mg的碱性氧化铝装填在固相萃取小柱中,取3 mL的二乙烯基苯混合物过柱,去除阻聚剂,将纯化后的二乙烯基苯低温避光储存。
1.2.2 生物炭表面改性 首先研磨稻壳生物炭,过70目筛。取20.0 g生物炭溶于200 mL氢氧化钠溶液(3 mol/L)中,在60 ℃下搅拌2 h。用G5砂芯漏斗分离得到的碱性处理的生物炭颗粒,用乙醇和蒸馏水洗涤,直到洗脱液呈中性。随后将碱性处理后的生物炭在-0.1 MPa和60 ℃下真空干燥8 h,干燥避光保存。
1.2.3 四氧化三铁的制备 将1.99 g FeCl2·4H2O和3.24 g FeCl3溶于100 mL蒸馏水中。配制50 mL 2 mol/L的NaOH溶液,80 ℃水浴加热,之后在搅拌状态下逐滴加入5 mL铁离子溶液,继续剧烈搅拌30 min。通过外加磁场分离收集产物,用蒸馏水洗涤至pH为中性,然后将制备的产物Fe3O4在真空中干燥。
1.2.4 皮克林乳液聚合法制备MMIPMs 以生物炭和Fe3O4作为皮克林乳液共稳定剂,形成水包油型乳液体系,其中水相为蒸馏水,油相为甲苯。取90 mg生物炭和30 mg Fe3O4,将其分散在12 mL蒸馏水中,超声处理10 min。取1 454 μL甲苯置于离心管中,加入200 μL 48 mg/mL的TC甲醇溶液、64 μL甲基丙烯酸、278 μL二乙烯基苯、30 mg偶氮二异丁腈,混匀作为油相。然后将油相与水相混合,手动摇晃3 min,直至形成稳定的皮克林乳液,60 ℃水浴下反应5 h。聚合反应结束后,将乳液倒入G5砂芯漏斗中抽滤,用甲醇洗涤3次,除去残留的低聚物和单体。利用甲醇—乙酸(体积比9∶1)洗涤,去除模板分子TC,真空干燥得到最终的MMIPMs。
非印迹材料(MNIPMs)的合成除不加模板分子TC外与上述印迹材料合成方法一致。
1.2.5 材料的表征
(1) 扫描电镜:将合成的材料粘在导电胶上喷金处理通过扫描电镜观察微球形貌。
(2) 红外光谱:干燥的待测样品与干燥的溴化钾按1∶100的比例混合,于研钵中研磨压片,用傅里叶变换红外光谱进行分析测试。
(3) X射线衍射:在2θ为10°~90°范围内对Fe3O4和MMIPMs的晶型结构进行表征。
(4) 热重分析:以10 ℃/min的加热速率,在45~600 ℃范围内检测材料的热稳定性。
1.2.6 材料的吸附性能
(1) 吸附动力学试验:称取10 mg MMIPMs/MNIPMs材料,分别加入5 mL 20 mg/L的TC甲醇溶液,分散于棕色容量瓶中。放置于摇床上振荡,通过磁分离取振荡不同时间后(5,10,20,40,60,120,240 min)的样品上清液。通过紫外分光光度计测定上清液吸光度,并按式(1)计算吸附容量。
(1)
式中:
Q——吸附容量,mg/g;
Ci——溶液中模板分子吸附前的浓度,mg/L;
Cf——溶液中模板分子吸附后的浓度,mg/L;
V——样品溶液的体积,L;
m——合成材料的质量,g。
(2) 等温吸附性能评价:称取10 mg MMIPMs/MNIPMs置于棕色容量瓶中,分别加入5 mL不同浓度(5,10,20,40,60,80 mg/L)TC溶液。振荡一段时间后,通过磁分离手段取上清液。通过紫外分光光度计测定上清液吸光度,并按式(1)计算吸附容量。
(3) 选择吸附试验:TC和OTC作为结构类似对照组,SMZ和TMC作为参比化合物对照组,进行选择性试验。首先,称取10 mg MMIPMs和MNIPMs加入到棕色容量瓶中,配置浓度为40 mg/L不同物质的甲醇标准溶液,分别取5 mL上述溶液加入到容量瓶中。放置在摇床上,室温下振荡混合物一段时间后,用外加磁场分离混合物,上清液通过尼龙66膜(0.22 μm)过滤。用紫外分光光度计分别检测4种分析物的浓度,按式(1)计算吸附容量,根据式(2)~(3)计算分配系数、选择性系数和相对选择性系数。
(2)
(3)
(4)
式中:
Kd——分配系数;
K——选择性系数;
K’——相对选择性系数。
1.2.7 固相萃取与HPLC联用
(1) 样品处理:5.0 g鸡肉/鱼肉样品,加入5 mL Na2EDTA-MCIlvaine缓冲液,冰水浴中超声处理20 min,然后8 000 r/min离心10 min,重复操作两次,合并两次上清液。水样取5 mL。
(2) 磁性固相萃取程序:称取20.0 g MMIPMs,加入上述样品提取液,振荡提取2 h,通过外加磁铁分离,丢弃上清液。用3 mL超纯水—甲酸(体积比80∶20)洗脱20 min,磁分离取上清液,过0.22 μm滤膜,然后用HPLC进行测定。
(3) HPLC条件:色谱柱为Xterra C18反相色谱柱(100 mm×2.1 mm,5 μm);流动相为含30%甲醇的乙腈溶液—0.01 mol/L柠檬酸溶液(体积比21∶79);流速1.0 mL/min;柱温35 ℃;进样量20 μL;检测波长357 nm。
2 结果与分析
2.1 结构表征
图1(a)表明,通过皮克林乳液聚合法合成的MMIPMs为球形结构,生物炭和Fe3O4分散附着在球体表面。通过对合成的MMIPMs和MNIPMs进行傅里叶红外光谱分析,验证磁性生物炭微球的成功制备。如图1(b)所示,3 448 cm-1处为生物炭分子间氢键O—H的伸缩振动,2 927 cm-1处为生物炭中脂肪烃或环烷烃中C—H的伸缩振动,1 737 cm-1处为甲基丙烯酸的C═O吸收峰,1 603~1 447 cm-1的4个强度不等的峰为二乙烯基苯的苯环C═C骨架振动,575 cm-1为Fe—O的吸收峰,上述结果表明成功合成了MMIPMs。通过X射线衍射仪进一步分析所制备的Fe3O4和MMIPMs的磁特性,如图1(c)所示,合成的Fe3O4在2θ(30.24°,35.59°,43.25°,57.29°,62.88°)处有衍射峰,分别为(220)、(311)、(400)、(511)和(440)面的特征衍射峰,与反尖晶石型的Fe3O4标准衍射谱的特征衍射图吻合较好(JCPDS card, No. 19-0629)。此外,MMIPMs的衍射图谱与Fe3O4的峰位相似,表明在MMIPMs合成过程中Fe3O4的结晶结构得到了很好的保持,因此合成的MMIPMs具有Fe3O4超顺磁性,可以应用于磁分离。通过热重分析仪对MMIPMs的热稳定性进行了分析,结果如图1(d)所示。在温度达到380 ℃之前,材料基本无质量损失;380~458 ℃有明显的失重(60%),在此阶段主要是由于聚合物基本解体,主体骨架坍塌由MMIPMs的热降解所致,剩余的组分为生物炭和Fe3O4粒子。以上结果表明,通过皮克林乳液聚合法,只需要简单地将Fe3O4作为共稳定剂,便成功制备了形状规则的MMIPMs,而且合成的材料具有很好的热稳定性。
2.2 材料的吸附性能评价
2.2.1 吸附动力学 吸附动力学是评价印迹材料吸附效率的重要指标。如图2(a)所示,MMIPMs和MNIPMs对TC的吸附速率呈先增加后减小的趋势,在吸附初始阶段聚合物表面暴露足够多的结合位点,而随着吸附的进行,大多数结合位点被TC占据,导致后续吸附过程中的空间阻力较大,吸附速率下降。另外,聚合物在吸附30 min内就可达到最大吸附量的50%,吸附60 min后吸附量呈现极缓慢的增长,直至120 min后吸附达到平衡,吸附平衡时间短,是因为生物炭的多孔结构提高了传质速率。在实际应用过程中,快的传质速率有利于增强前处理过程中的萃取效率。
2.2.2 等温吸附曲线 如图2(b)所示,MMIPMs和MNIPMs
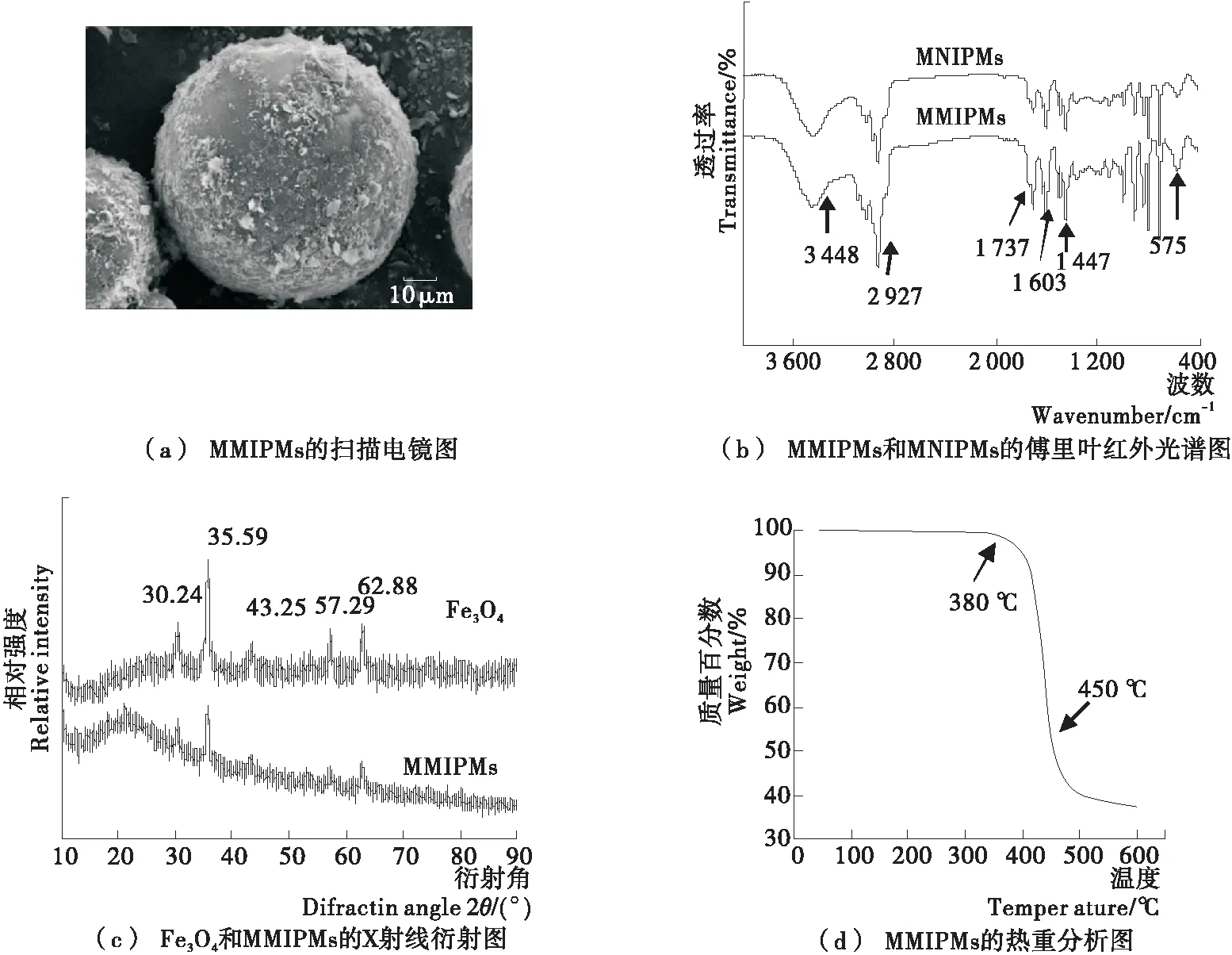
图1 聚合物的扫描电镜、傅里叶红外光谱、X射线衍射和热重分析表征
Figure 1 Characterizations of polymers by scanning electron microscope, fourier transform infrared spectrometer,
X-ray diffractometer and thermogravimetric analysis
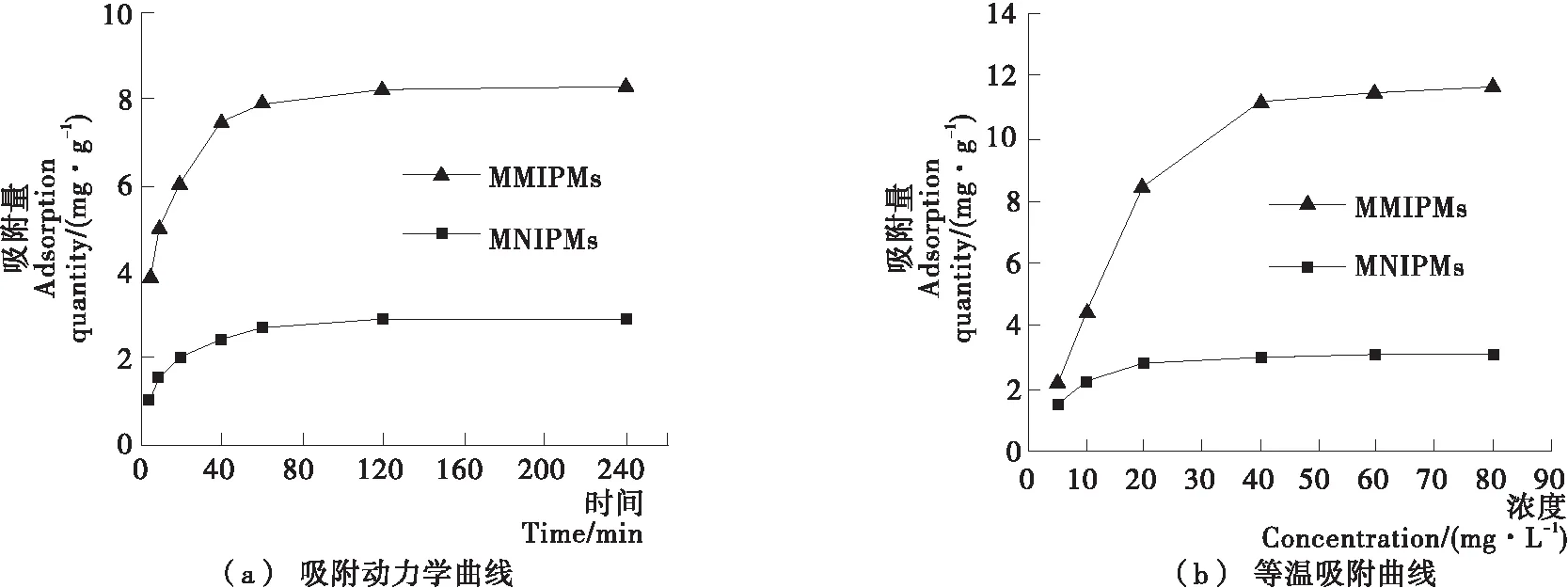
图2 MMIPMs和MNIPMs的吸附动力学曲线以及吸附等温线
的吸附量随初始浓度的增加而增加。与MNIPMs相比,MMIPMs明显具有更高的吸附能力,当非印迹材料达到吸附饱和时,印迹材料吸附量仍呈上升趋势。当溶液初始浓度为80 mg/L时,印迹材料和非印迹材料的吸附量分别为11.68,3.08 mg/g,印迹因子为3.79,说明MMIPMs具有较好的印迹效果。
2.2.3 选择性试验 由表1可知,MMIPMs对TC的吸附量明显高于对其他分析物(OTC、SMZ、TMC)的吸附量,且高于MNIPMs的吸附量。MMIPMs对TC的吸附量(11.1 mg/g)是对SMZ吸附量(0.353 mg/g)的31.4倍,表明制备的MMIPMs对模板分子TC表现出良好的选择性。用选择性系数K进一步评价MMIPMs和MNIPMs的吸附选择性。显然,对于4种参比物质,MMIPMs的选择性系数均大于MNIPMs,且MMIPMs和MNIPMs的相对选择性系数K’>1,进一步证实了MMIPMs对TCs的良好特异性识别能力。
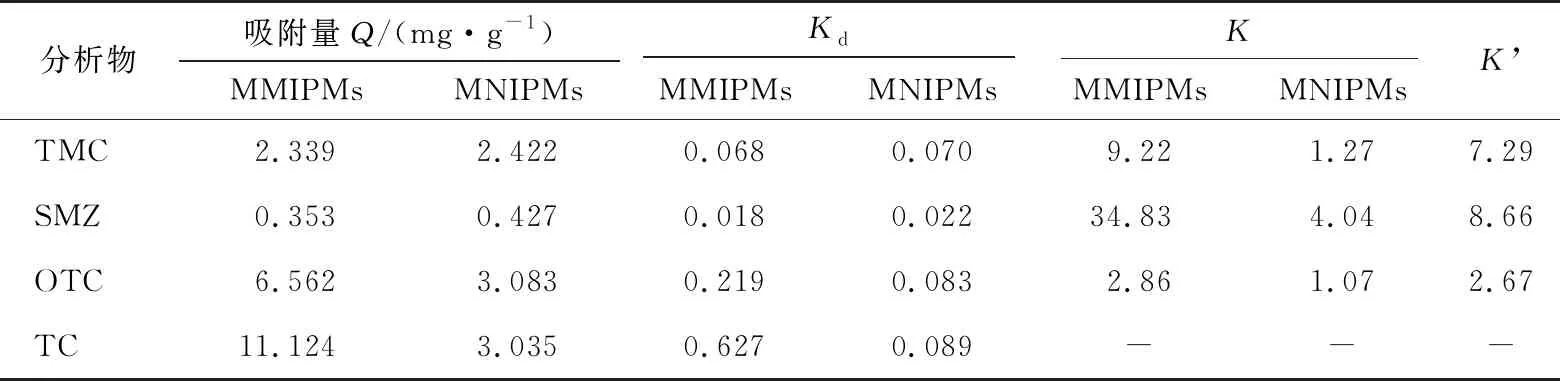
表1 MMIPMs和MNIPMs对4种分析物的选择性
2.3 固相萃取条件的优化
为了得到最佳的萃取效果,对试验过程中可能影响萃取效率的因素进行优化,包括洗脱溶剂的种类、洗脱剂体积和洗脱时间,结果见图3。选用不同种类的洗脱液(10%~20%甲酸乙腈溶液、5%~20%甲酸水溶液)对吸附后的MMIPMs淋洗,如图3(a)所示,当洗脱液为20%的甲酸水溶液时,获得的回收率最高(82.4%~85.8%),因此选用20%的甲酸水溶液洗脱。选用不同体积(2~5 mL)的20%甲酸水溶液进行洗脱,如图3(b)显示洗脱剂体积为3 mL时,3种TCs的回收率就能达到94.8%~104.0%。图3(c)为不同洗脱时间(10~60 min)下的回收率情况,当洗脱时间为20 min时,回收率为97.0%~100.0%),随着洗脱时间的增大,回收率没有明显的增长,因此选用20 min为洗脱时间。
根据优化试验结果,用3 mL含20%甲酸的水溶液作为洗脱剂,洗脱20 min进行后续研究。
2.4 实际样品中的应用
以合成的MMIPMs作为磁性固相萃取吸附材料,结合HPLC建立了一种适用于食品中TCs兽药残留的检测方法。在最佳萃取条件下,当TCs浓度为5~160 μg/L时,目标物浓度与峰面积之间呈现良好的线性关系(R2>0.997 2),选取信噪比(S/N)为3时的溶液浓度为TCs的最低检测限,TC和OTC仪器检测限分别为1.42,1.58 μg/L,方法检出限分别为0.46,0.51 μg/L。
为进一步证明所建方法的可行性,对鱼肉、鸡肉和水样品进行3个浓度水平加标(5,10,20 μg/kg),通过得到的加标回收率评价所建立方法的准确度。由表2可知,加标鱼肉、鸡肉、水样中的回收率分别为91.2%~103.4%,90.6%~98.9%,92.0%~101.4%,RSD为1.23%~9.99%,该结果满足GB/T 27404—2008定量分析检测要求(被测组分含量<0.1 mg/kg,回收率范围为60%~120%),与其他方法(表3)比较,试验方法具有较高的回收率和较低的检出限,说明该方法可用于食品中TCs残留定量分析。
3 结论
以生物炭和四氧化三铁作为皮克林乳液的共稳定剂,结合分子印迹技术,合成了一种新型磁响应四环素印迹生物炭微球。该方法制备过程简单、成本低、合成的聚合物为具有磁响应特性的规整球形结构,可以简化样品前处理的提取和净化环节。吸附性能研究表明得到的磁响应四环素印迹生物炭微球传质速率较快(120 min基本达到吸附平衡)、吸附量较大(11.68 mg/g)、对目标物具有良好的选择性(印迹因子为3.79)。以高效液相色谱仪作为分析仪器,所建立的检测方法实现了对食品中四环素类抗生素的准确分析(3种样品的回收率为90.6%~103.4%,RSD为1.23%~9.99%)。为生物炭在食品样品中污染物检测方面的应用提供了一条新的思路和方法。
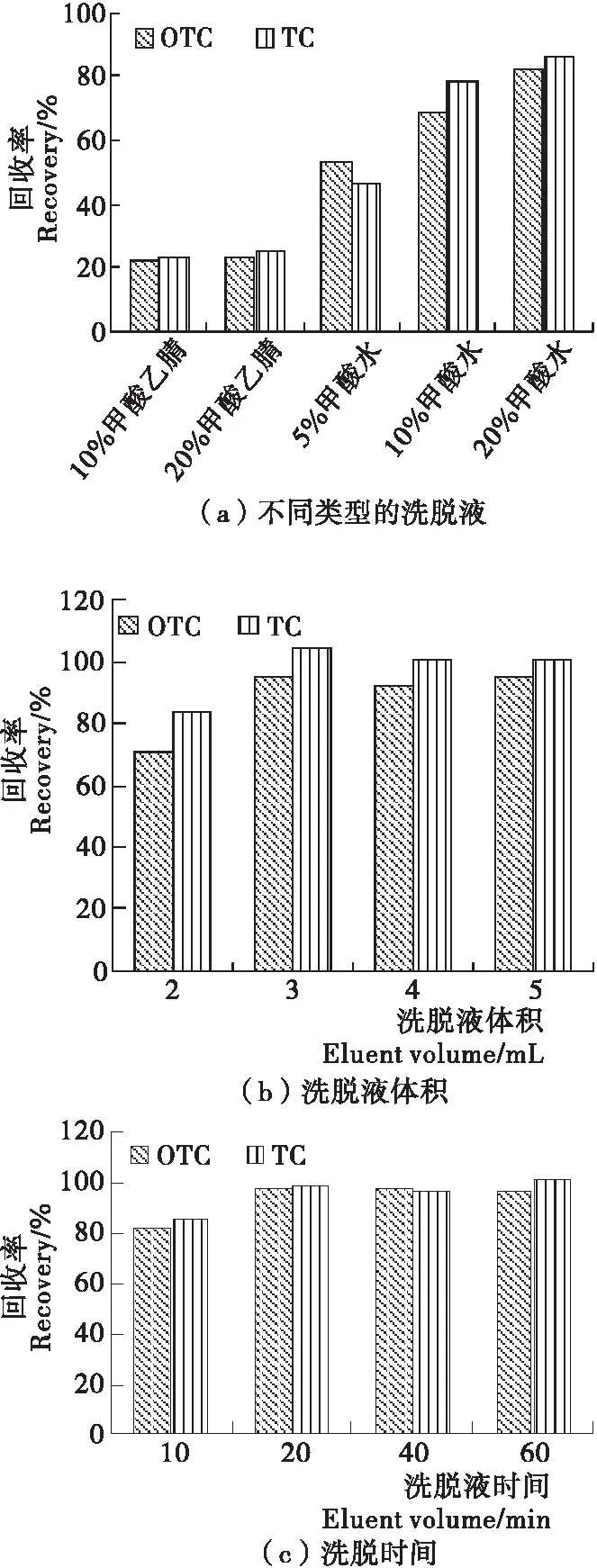
图3 不同萃取条件对回收率的影响
表2 不同样品中3种TCs的加标回收率及精密度
Table 2 The recoveries and RSDs of three TCs in different spiked samples (n=3)

TCs浓度/(μg·L-1)分析物 鱼肉/% 鸡肉/% 自来水/% 回收率RSD回收率RSD回收率RSD5OTC96.15.3598.24.94101.46.42TC103.43.8091.86.2892.01.2310OTC100.4 6.2598.95.88 95.71.56TC94.64.4392.53.7194.75.9920OTC91.21.3096.61.98 95.29.99TC97.11.7290.64.9593.65.51
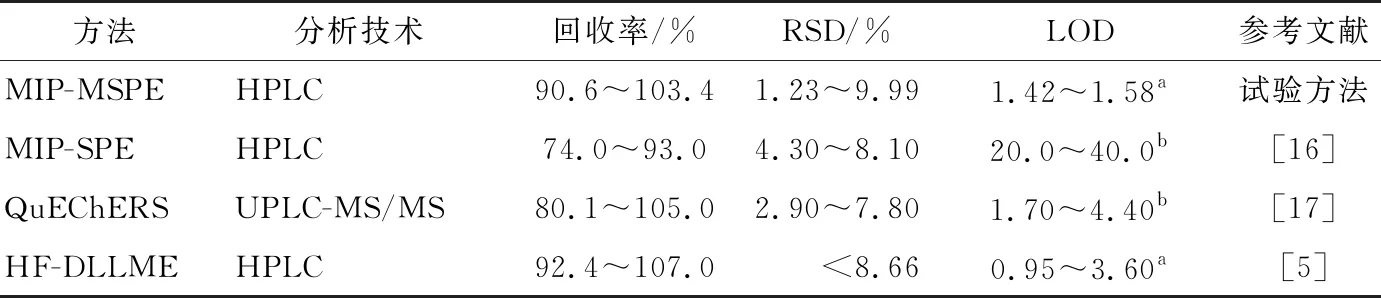
表3 与其他文献方法的比较†
† a 单位为μg/L;b 单位为μg/kg。
