利用信息肽诱导和Cre/LoxP系统构建猪链球菌基因缺失株
2019-05-10陈福广黄萌萌朱金鲁孟庆文刘思国刘建华张跃灵
刘 冉,陈福广,陈 平,黄萌萌,朱金鲁,谢 芳,孟庆文,刘思国,刘建华,张跃灵*
(1.新疆农业大学动物医学院,新疆 乌鲁木齐 8300522;2.中国农业科学院哈尔滨兽医研究所兽医生物技术国家重点实验室,黑龙江 哈尔滨 150069)
链球菌中存在调控自然感受态的ComR/S二元调控系统[5]。comS编码的多肽ComS在胞外被加工成7~9个氨基酸的信息肽XIP(SigX-inducing peptide),之后XIP进入胞内,结合并激活调控因子ComR,ComR激活后结合SigX的基因启动子并启动其表达,产生SigX启动感受态相关基因的表达,从而吸收并重组外来DNA[6-8]。基于该原理,研究者已将XIP诱导外源DNA的转化和重组应用于S.pneumoniae和S.mutans基因缺失株的构建[9-10]。但是,不同链球菌的ComS和XIP序列各不相同,具有很强的种特异性[5]。Zaccaria等证实S.suis2型05ZYH33菌株的 comS基因产物 GE9(序列 GNWGTWVEE)能够有效诱导05ZYH33菌株吸收外源质粒,而且还能够诱导其吸收DNA片段并发生重组,为S.suis基因缺失株构建提供了新的思路[11]。但是,目前还没有将这一技术用于S.suis基因缺失株构建的报道,原因可能是这种方法需要插入抗性基因进行筛选,而限于抗性基因可选数量有限,不利于后续的回复突变和多基因缺失。针对去除抗性基因这一问题,Koczula等利用了Cre/LoxP系统[12]。这一系统中Cre为噬菌体P1的重组酶,编码343个氨基酸组成的蛋白,识别特异的DNA序列,即LoxP位点,使LoxP位点间的基因序列被删除[13]。研究者尝试了多种LoxP位点,目前使用最多的是Lox66和Lox71位点,这两个位点被Cre酶识别并重组为Cre酶难以识别的Lox72位点[14]。Koczula等通过在质粒构建过程中引入LoxP位点,后期筛选获得基因缺失克隆后,再引入表达Cre重组酶的温敏质粒,切除LoxP位点之间的抗性基因,从而实现去除抗性基因的无痕缺失[15]。
本研究通过整合上述两种方法,将XIP诱导S.suisDNA片段转化重组应用于S.suis2型05ZYH33的基因缺失株构建中,以S.suis05ZYH33的α-甘露糖苷酶基因ssu05_1921为例,实现快速、高效构建抗性替代的单基因缺失株,同时也可去除抗性基因,进行多基因缺失,为构建S.suis基因缺失株,研究S.suis致病机制提供新的选择。
1 材料与方法
1.1 主要实验材料S.suis2型05ZYH33菌株,为中国2005年四川流行分离株[16]。大肠杆菌MC1061F-感受态购自上海唯地生物技术有限公司。THB培养基和LB培养基购自美国BD公司;THB(SPC),LB(Cm)和THB(Cm)培养基为本实验室自制。pSET4s、pSET6s由DaisukeTakamatsu教授惠赠[17]。PrimeSTAR Max DNA聚合酶和One Step SYBR PrimeScript RT-PCR Kit II购自宝生物工程(大连)有限公司;PCR产物纯化试剂盒和DNA胶回收试剂盒购自OMEGA公司;细菌基因组DNA提取试剂盒和质粒小提试剂盒购自TIANGEN公司;限制性内切酶、T4 DNA连接酶和碱性磷酸酶均购自Thermo Scientific公司;RNA Plus Mini Kit购自Qiagen公司。
1.2 信息肽GE9及引物的设计与合成 参考文献[11]中信息肽GE9序列(GNWGTWVEE),由哈尔滨博仕生物公司合成,纯度97%,溶于DMSO制成5 mmol/L溶液。根据GenBank中S.suis05ZYH33基因组(CP000407)、pSET2(AB055650)和 Cre重组酶基因(X03453)序列,利用Premer Premier 5.0软件设计引物(表1),由哈尔滨博仕生物公司合成。
1.3 ssu05_1921基因上、下游片段及spc基因的扩增和融合 采用细菌基因组DNA提取试剂盒提取S.suis05ZYH33的基因组DNA,采用质粒小提试剂盒提取pSET4s质粒。如图1的Step 1所示,以1921upF和1921upSalIR、1921dnXhoIF和1921dnR为引物,以S.suis05ZYH33基因组DNA为模板,分别扩增ssu05_1921基因的上游片段1921UP(1 363 bp,3'末端引入SalⅠ位点)和下游片段1921DN(1 312 bp,5'末端引入XhoⅠ位点)。以pSET4s为模板,以SpcloxSalIF和SpcloxXhoIR为引物,扩增获得Lox66-spc-Lox71片段(1 295 bp),其5'末端引入Lox66位点(TACCGTTCGTATAATGTATGCTATACGAAGTT AT)和SalⅠ酶切位点,3'末端引入Lox71(TACCG TTCGTATAGCATACATTATACGAAGTTAT)和XhoⅠ酶切位点。分别胶回收上述1921UP、1921DN和Lox66-spc-Lox71等3个DNA片段的PCR产物,以摩尔比1∶1∶1混合,SalⅠ和XhoⅠ双酶切,酶切产物纯化回收后以T4 DNA连接酶连接。以连接产物为模板,以1921upF和1921dnR为引物,PCR扩增3个DNA片段融合产物1921UP-Lox66-spc-Lox71-1921DN。

表1 实验所用引物信息Table 1 Primer information used in this experiment
1.4S.suis05ZYH33 ssu05_1921基因缺失株的构建 挑取THB平板上S.suis05ZYH33单菌落,接种THB液体培养基,37℃,5%CO2静置培养过夜,第2 d 1∶100(v/v)转接培养,分别于培养1 h、1.5 h、2 h、2.5 h时取100μL加入转化管(转化管中为提前5 min加入的5 mmol/L GE9和1921UP-Lox66-spc-Lox71-1921DN片段),混匀后,37℃静置培养2 h,涂布THB(SPC)平板过夜培养。挑取10个单菌落,接种 THB液体培养基。培养至 OD600nm>0.4时以1921cfmF和1922cfmR为引物,进行菌液PCR鉴定阳性克隆并测序。
1.5 表达Cre重组酶的pSET6s/PtufA-cre载体的构建 以S.suis05ZYH33基因组DNA为模板,以tufAproPstIF和tufAproSalIR为引物,扩增tufA基因的上游启动子片段PtufA;由华大基因公司合成重组酶cre基因(X03453),以cre基因为模板,以Cre-SalIF和CrePstIR为引物,扩增获得cre基因片段。胶回收PtufA和cre片段后,利用1.3中片段重组方法,经SalⅠ酶切后PCR重组获得PtufA-cre融合片段。将PtufA-cre片段PstⅠ单酶切,与经PstⅠ单酶切并经碱性磷酸酶处理的pSET6s质粒连接,转化大肠杆菌MC1061F-,经LB(Cm)平板筛选,挑取单克隆培养于LB(Cm)液体培养基,提取质粒进行测序验证。
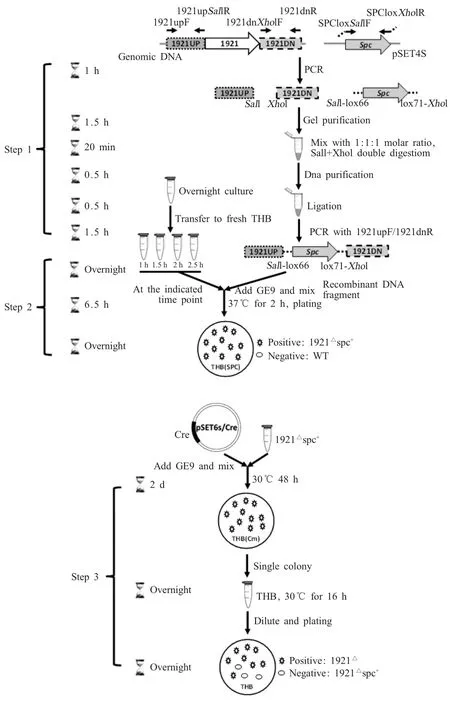
图1 S.suis 05ZYH33基因缺失株构建流程示意图Fig.1 Diagram of the construction of S.suis 05ZYH33 gene-deleted strain
1.6 spc抗性基因的敲除 制备pSET6s/PtufA-cre质粒,转化1921Δspc+菌株,按1.4转化方法进行,仅将1.4中的重组片段改为pSET6s/PtufA-cre质粒。转化后涂布THB(Cm)平板,30℃培养48 h后挑取单菌落接种THB液体培养基,30℃过夜培养使Cre表达并作用。第2 d稀释涂THB平板,37℃培养并挑取10个单菌落接种THB液体培养基,37℃过夜培养使温敏质粒pSET6s/PtufA-cre丢失。采用spc基因扩增引物SpcloxSalIF和SpcloxXhoIR进行菌液PCR鉴定并测序,通过划线SPC及Cm平板进行抗性鉴定。
1.7 RT-PCR检测S.suis05ZYH33 ssu05_1921基因缺失及相邻基因转录 采用RNA Plus Mini Kit提取S.suis05ZYH33野生菌株(WT)和缺失菌株的总RNA,测定RNA浓度并鉴定RNA质量,采用表1中的引物,按One Step SYBR PrimeScript RT-PCR Kit II方法进行RT-PCR,设看家基因secA(ssu05_1814)[18]为参考基因,验证ssu05_1921基因的缺失,并检测上下游相邻基因ssu05_1920和ssu05_1922的转录情况。
十九岁,别呦呦遇上了他。谈到他,别呦呦脸色微红,欲羞还笑。她没说他叫什么名字,也没说他有多大,只说他“有男人的味道”,“他是属蜘蛛的,织了一张网,我是一只刚从荷叶上飞起来的蜻蜓,眼前还是绿水,鼻中还有幽香,看见网,一头撞了进去”。
2 结果
2.1ssu05_1921基因上、下游片段及spc基因的扩增和融合 经PCR鉴定ssu05_1921基因上、下游片段及spc基因的扩增和融合,结果显示1921UP、1921DN、Lox66-spc-Lox71及 1921UP-Lox66-spc-Lox71-1921DN片段大小与预期符合,表明1921UP-Lox66-spc-Lox71-1921DN正确融合(图2)。

图2 1921UP、Lox66-spc-Lox71和1921DN的PCR扩增和融合Fig.2 Amplification and fusion of the 1921UP,Lox66-spc-Lox71 and 1921DN sequence
2.2S.suis05ZYH33ssu05_1921基因缺失株1921Δspc+的构建 经菌液PCR鉴定ssu05_1921基因缺失株的构建,扩增鉴定从上游同源臂上游198 bp至下游同源臂下游220 bp的产物,结果显示10个克隆扩增片段均为4.4 kb,小于野生菌株扩增片段5.2 kb,与预期符合(图3),经测序分析重组位置正确,表明正确构建1921Δspc+菌株。
2.3 表达Cre重组酶的pSET6s/PtufA-cre载体的构建经PCR鉴定两片段的扩增和融合,结果显示PtufA,cre及融合片段PtufA-cre大小均与预期符合(图4A)。酶切鉴定pSET6s/PtufA-cre(图4B)并测序结果与预期一致,表明正确构建了重组载体pSET6s/PtufA-cre。

图3 1921Δspc+缺失株的PCR鉴定Fig.3 Identification of 1921Δspc+strains by PCR

图4 两片段及融合片段扩增和重组质粒酶切鉴定Fig.4 Identifications of the DNA fragments and the recombinant plasmid by PCR(A)and restriction enzyme digestion(B),respectively
2.4 spc抗性基因敲除鉴定结果 经菌液PCR鉴定spc抗性基因的去除,结果显示10个克隆中有4个spc基因去除(图 5);经 THB、THB(SPC)和 THB(Cm)平板培养,结果显示这4个不能在THB(SPC)平板上生长(图6),表明spc基因被去除,且这10个克隆均不能够在THB(Cm)平板生长,表明升高温度将温敏质粒pSET6s/PtufA-cre丢失。
测序分析结果显示spc抗性基因被去除,且位置序列正确,再次表明获得了S.suis05ZYH33的无抗性标记的基因缺失株1921Δ。
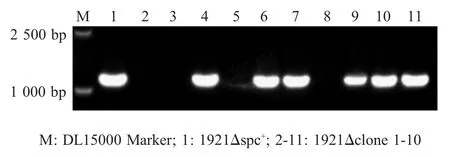
图5 10个随机克隆中spc基因的PCR检测Fig.5 Identification of spc gene in 10 random ly chozen clones by PCR

图6 10个随机克隆的抗性检测Fig.6 Resistance detection of 10 random ly chosen clones
2.5 RT-PCR检测S.suis05ZYH33 ssu05_1921基因缺失及相邻基因转录 利用One Step SYBR PrimeScript RT-PCR Kit II对各菌株进行鉴定,结果显示WT菌株中ssu05_1921基因正常表达,而1921Δ 菌株中无ssu05_1921基因的表达(图 7,2),相邻基因ssu05_1920和ssu05_1922在WT菌株和1921Δ菌株的表达水平一致(图 7,1和3),表明ssu05_1921基因已缺失,且相邻基因的转录未受到ssu05_1921基因缺失的影响。

图7 ssu05_1921及相邻基因的RT-PCR检测Fig.7 Detection of the expression of ssu05_1921 and its neighbor genes by RT-PCR
3 讨论
构建基因缺失株是发掘致病因子、研究致病因子功能和阐释致病机制的重要研究手段。相比其它链球菌,S.suis基因缺失株的构建技术还不够完善,限制了其致病机制的研究。目前S.suis缺失株构建多采用电转化温敏质粒pSET4s的方法,将重组片段构建至pSET4s后电转化S.suis,经过第一次同源重组筛选和第二次同源重组筛选,获得目的基因缺失菌株[10-11]。这种方法对其感受态制备和转化设备及技术要求较高。更重要的是二次同源重组的效率低,据报道仅为2.6%[17],而且是无抗性筛选,常常经过大量繁琐的筛选,仍然难以获得目的缺失株。另外,二次同源重组的筛选需要多次传代,费时长,而且使病原细菌自发毒力降低,对致病机制的研究十分不利[19]。
本研究探索整合了调控自然感受态的ComR/S系统和Cre/LoxP系统,利用GE9诱导S.suisDNA片段转化重组,构建S.suis基因缺失株。在验证基因缺失方面,首先采用上游同源臂上游和下游同源臂下游特异引物,从而保证扩增产物来自基因组上ssu05_1921基因的位置,测序证实该位置先发生了spc抗性基因替代ssu05_1921基因后经Cre酶作用,spc基因被去除,而且位置和序列正确,初步证明本研究建立的基因缺失方法可行,基因能够按照预期发生替代和缺失。采用RT-PCR,确认ssu05_1921基因在野生菌株中表达,在ssu05_1921缺失菌株中不再表达,进一步证实ssu05_1921基因缺失,说明本研究建立的基因缺失方法切实可行。本研究也尝试进行western blot和α-甘露糖苷酶功能验证,但是S.suis05ZYH33野生菌株在实验条件下western blot无法检测到其蛋白,也检测不到α-甘露糖苷酶活性,限制了western blot和α-甘露糖苷酶功能验证。肺炎链球菌的α-甘露糖苷酶SpGH92也存在相同的情况,研究者推测SpGH92表达量少可能受到某种调控[20]。本研究构建的ssu05_1921基因缺失株为进一步研究ssu05_1921基因产物在猪链球菌中的功能和调控奠定了基础。
相比电转化温敏质粒的方法,该方法无需制备电转感受态和电转化,对技术和设备要求低,操作简便。只需单次转化和传代,2 d即可快速构建含有抗性基因的单基因缺失株,用于单基因功能研究。由于采用抗性筛选,效率可接近100%。该方法在单基因功能研究方面具有强大优势。
在需要多基因缺失、抗性基因选择有限时,再引入表达Cre重组酶的温敏质粒,切除LoxP位点之间的抗性基因,实现无痕缺失,解除后顾之忧。该步骤约需4 d,传代2~3次,抗性基因去除率高(本文例子中抗性基因去除率约40%)。相比多次传代筛选二次同源重组,重组率2.6%的pSET4s方法也具有明显的优势。本研究建立的这种高效的S.suis基因缺失方法将在发掘S.suis致病因子,研究S.suis致病机制方面发挥重要作用。
