基于高通量测序技术对沙湖水体细菌多样性的分析
2017-12-19张燕灵李靖宇刘建利刘雅琴张翼飞
张燕灵,李靖宇,刘建利,刘雅琴,张翼飞,赵 吉,邓 蔚
(1. 北方民族大学 生物科学与工程学院, 银川 750021; 2. 内蒙古自治区环境科学研究院, 呼和浩特 010010; 3. 内蒙古大学 环境与资源学院, 呼和浩特 010021)
基于高通量测序技术对沙湖水体细菌多样性的分析
张燕灵1,李靖宇1,刘建利1,刘雅琴1,张翼飞2,赵 吉3,邓 蔚1
(1. 北方民族大学 生物科学与工程学院, 银川 750021; 2. 内蒙古自治区环境科学研究院, 呼和浩特 010010; 3. 内蒙古大学 环境与资源学院, 呼和浩特 010021)
为了解沙湖水体中细菌群落结构和多样性,采用E.Z.N.A.®水样DNA提取试剂盒提取水样总DNA,对细菌群落的16S rRNA基因的V4~V5区进行MiSeq测序,利用Mothur软件和R语言进行细菌群落结构及多样性的分析。共得到427 975条优化序列,在97%的相似水平下对OTU进行统计分析,得到20门,50纲,95目,161科,276属。样点A、B、C、D的OTU分别为497、496、487和511。优势类群为变形菌门(Proteobacteria)、蓝细菌门(Cyanobacteria)、放线菌(Actinobacteria)、拟杆菌(Bacteroidetes)。沙湖水体中细菌群落结构复杂,但不同位点细菌群落结构的组成以及多样性差异不显著。
细菌群落;沙湖;水体;MiSeq测序
细菌作为湖泊生态系统的重要组成部分,在湖泊的物质循环和能量流动中起着重要作用。由于传统纯培养技术限制,利用平板培养法测定的细菌类群数量只能占到湖泊中实际存在的细菌总数的1%~10%[1]。近年来,高通量测序技术的发展,为细菌多样性研究开辟了新的途径,不断革新着我们对细菌多样性的认识。高通量测序可获得的基因序列以百万甚至亿万计,不但分辨率高,而且可以同时分析上百个不同的样品,是分析自然界中细菌群落结构组成和相对丰度的重要工具[2]。目前,许多学者对宁夏沙湖的浮游植物和浮游动物进行了研究报道[3-5],但是关于沙湖水体的细菌群落结构和多样性的研究还未见报道。据此,本研究通过高通量测序技术对宁夏沙湖细菌群落开展研究,从而了解沙湖不同位点的细菌群落的结构、丰度和多样性,为深入认识湖泊的生物地球化学循环提供细菌生态学视角。
1 材料与方法
1.1 材料
供试水样取自宁夏回族自治区平罗县沙湖旅游区(38°47′N~38°50′N,106°20′E~106°23′E)。水体样品的采集时间为2015年10月2日,根据沙湖的形状选取12个取样点A1~A3,B1~B3,C1~C3,D1~D3(图1)。将水样运回实验室保存于4℃冰箱备用。取样点作图利用Landsat 8影像,2014年10月23日下载于美国地质调查局(www.usgs.gov),利用band5、band4、band3 3个波段合成假彩色影像图,然后和band8融合为分辨率为15 m的最终影像图。利用arcgis10.0,叠加取样点坐标即为取样示意图。

图1 沙湖采样点设定
1.2 方法
1.2.1 水样基因组总DNA提取及扩增
每个点样品取2.5 L水样后经0.22 μm滤膜(直径150 mm; Millipore)过滤,并将滤膜在无菌条件下剪碎。然后采用E.Z.N.A.®water DNA Kit试剂盒(Omega, USA),参照说明书提取沙湖水体细菌DNA。提取的DNA用1%琼脂糖凝胶电泳进行检测。
20 μL PCR反应体系:2.0 μL 2.5×10-3mol/L dNTPs,0.8 μL 0.5×10-5mol/L 338F (5′-ACT CCT ACG GGA GGC AGC AG-3′),0.8 μL 0.5×10-5mol/L 806R (5′-GGA CTA CHV GGG TWT CTA AT-3′) ,2.0 μL 10×缓冲液,0.2 μL BSA,10 ng模板以及0.2 μL,5 U/μLrTaq聚合酶,最后加ddH2O到20 μL。
反应条件:95℃预变性3 min;95℃变性30 s,55℃退火30 s,72℃延伸45 s,28个循环;72℃再延伸10 min。
获得的PCR产物使用AXYGEN公司的AxyPrep DNA凝胶回收试剂盒切胶回收;使用Promega公司的QuantiFluorTM-ST蓝色荧光定量系统进行定量并将每个样品按测序量比例混合,然后根据标准流程进行Illumina MiSeq双端测序。
1.2.2 Illumina MiSeq测序数据分析
利用Trimmomatic和FLASH软件,过滤read尾部质量值20以下的碱基,设置50 bp的窗口,如果窗口内的平均质量值低于20,从窗口开始截取后端碱基,过滤质控后50 bp以下的read;根据PE reads之间的overlap关系,将成对reads拼接(merge)成一条序列,最小overlap长度为10 bp;拼接序列的overlap区允许的最大错配比率为0.2,筛选不符合序列;根据序列首尾两端的barcode和引物区分样品,并调整序列方向,barcode允许的错配数为0,最大引物错配数为2。进一步运用RDP Classifier 软件包进行系统发育分析,在97%置信度下将每个样品中的所有16S rRNA基因序列进行分类,获得门、纲、目、科和属各水平下的分类单元。Alpha多样性分析采用mothur[7]version v.1.30.1指数分析,设置指数评估的OTU相似水平为97%,计算多样性指数:菌群丰度指数,Chao和Ace;菌群多样性指数,Shannon和Simpson;深度指数,Coverage。应用R语言进行PCA统计分析和作图,其中一个点代表一个样品,颜色和形状相同的点同属于一个组,两个点之间的距离越远表示两个样本组成差异越大。
2 结果与分析
2.1 测序数据分析
通过各样品中细菌的16S rRNA 基因 V4-V5 区测序,12个样品测得的高质量序列为427 975条,其中A1为37 018,A2为38 758,A3为31 082,B1为31 032,B2为32 579,B3为43 747,C1为34 558,C2为33 599,C3为38 466,D1为29 210,D2为39 143,D3为38 783。如图2所示,由Shannon-Wiener 指数结合测序深度绘制的12个样品曲线趋向平坦,说明测序数据量足够大,可以反映样本中绝大多数的细菌信息。
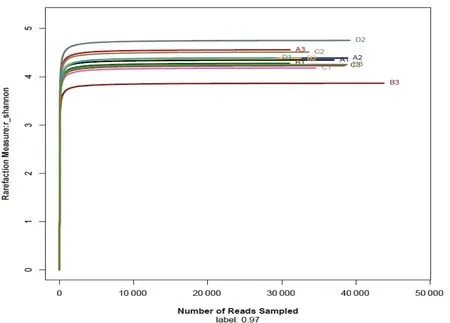
图2 样品Shannon-Wiener曲线图
2.2 沙湖水体细菌群落结构
由细菌样本聚类树与柱状图组合分析可知样本中细菌的组成、相对丰度及样本间基于群落组成的层次聚类[9]。如图3-A所示,在细菌分类学门水平上,高通量测序发现沙湖水样12个取样点拥有7个相同的优势细菌类群(丰度>0.7%为优势类群),它们分别是变形菌门(Proteobacteria)、蓝细菌门(Cyanobacteria)、放线菌(Actinobacteria)、拟杆菌(Bacteroidetes)、厚壁菌门(Firmicutes)、疣微菌门(Verrucomicrobia)、绿弯菌门(Chloroflexi )。
各取样点的细菌群落差异各不相同,这种差异体现在某些种群的相对丰度上,由图3-A可知,A1和A2的细菌群落结构最为相似,各细菌的相对丰度分别为:变形菌门,A1为32.13%,A2为29.72%;蓝细菌门,A1为15.38%,A2为22.09%;放线菌,A1为29.19%,A2为27.17%;拟杆菌,A1为12.19%,A2为11.1%;厚壁菌门,A1为4.72%,A2为3.51%;疣微菌门,A1为2.97%,A2为2.52%;绿弯菌门,A1为1.73%,A2为1.87%。对于相对丰度<0.7%的弱势细菌类群,A1中的细菌总量为1.68%,A2为1.75%。其次为B1和B2,各细菌的相对丰度分别为:变形菌门,B1为24.85%,B2为26.36%;蓝细菌门,B1为21.62%,B2为17.18%;放线菌,B1为23.95%,B2为28.4%;拟杆菌,B1为20.98%,B2为17.12%;厚壁菌门,B1为3.18%,B2为2.88%;疣微菌门,B1为2.66%,B2为4.1%;绿弯菌门,B1为1.09%,B2为2.2%。对于相对丰度<0.7%的弱势细菌类群,B1中的细菌总量为1.67%,B2为1.77%。而C2中的各种细菌相对丰度与其他取样点相差最大。C2中的各细菌相对丰度分别为:变形菌门为35.29%,蓝细菌门为27.47%,放线菌为7.77%,拟杆菌为19.88%,厚壁菌门为1.41%,疣微菌门为4.47%,绿弯菌门为2.63%,弱势细菌总量为1.09%。
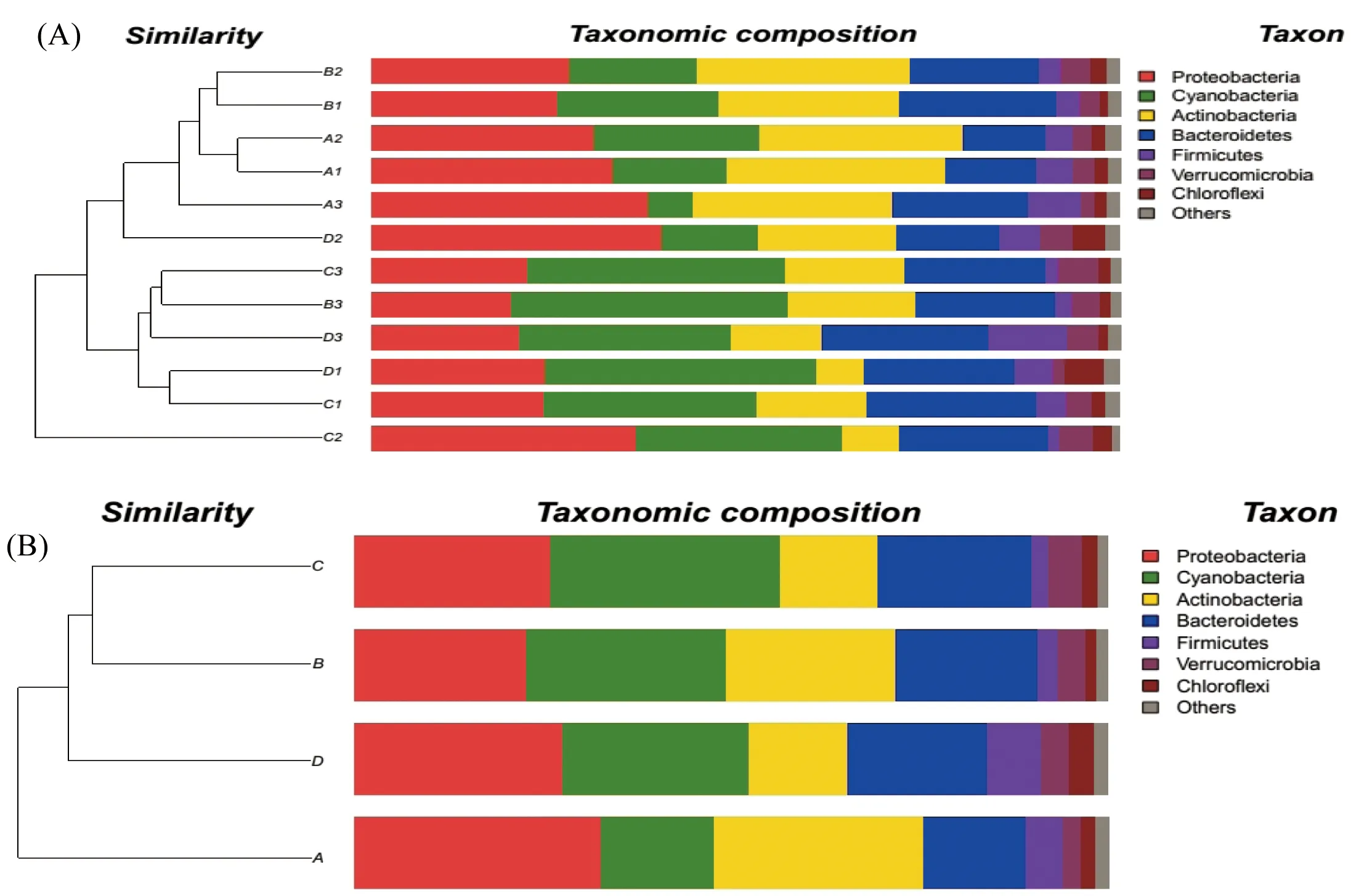
图3 沙湖水体细菌在门水平的高通量相对丰度及相似性
如图3-B所示,同一区域3个样点的测序数据看成一个整体,水样A、B、C和D的细菌群落最为相似的是C和B,其次为D,而A和其他的相差最大,但差异不显著。各细菌的相对丰度分别为:变形菌门,A为32.65%,B为22.77%,C为26.06%,D为27.61%;蓝细菌门,A为15.09%,B为26.56%,C为30.36%,D为24.78%;放线菌,A为27.71%,B为22.48%,C为12.96%,D为12.98%;拟杆菌,A为13.52%,B为18.82%,C为20.35%,D为18.46%;厚壁菌门,A为4.97%,B为2.67%,C为2.35%,D为7.17%;疣微菌门,A为2.45%,B为3.59%,C为4.47%,D为3.66%;绿弯菌门,A为1.8%,B为1.54%,C为2.02%,D为3.44%。劣势细菌总量分别为:A为1.82%,B为1.56%,C为1.43%,D为1.9%。
2.3 沙湖水体细菌多样性
如表1所示,在97%相似度下,A、B、C、D的ace指数分别为509、511、502和516,chao指数分别为514、512、509和521,Ace指数和Chao指数均表明取样点D的物种总数最多,而A、B、C、D的Shannon指数分别为4.47、4.24、4.46和4.62,Simpson指数分别为0.022、0.0383、0.0331和0.0243,Shannon指数表明取样点D的物种种类最多,而Simpson指数表明取样点A的物种种类最多。所有取样点的Coverage均大于99%,说明本次测序结果能代表样本的真实情况。
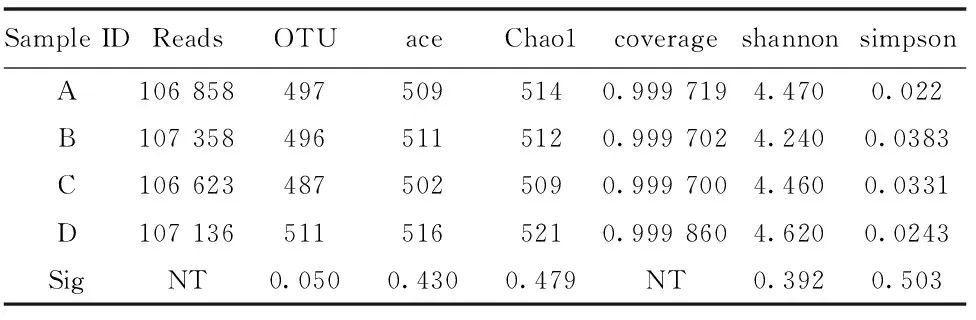
表1 不同样品细菌群落的丰度和多样性指数
注:NT表示没有检测不同样品之间的显著性;P<0.05表示显著
Rank-abundance[10]曲线是分析物种丰度和物种均匀度的一种方式,如图4所示,在97%相似度的OTU下,D的细菌丰度最高,其次为A,而C的细菌丰度最低。并且D的细菌种类分布最均匀,而C的细菌种类分布最不均匀。
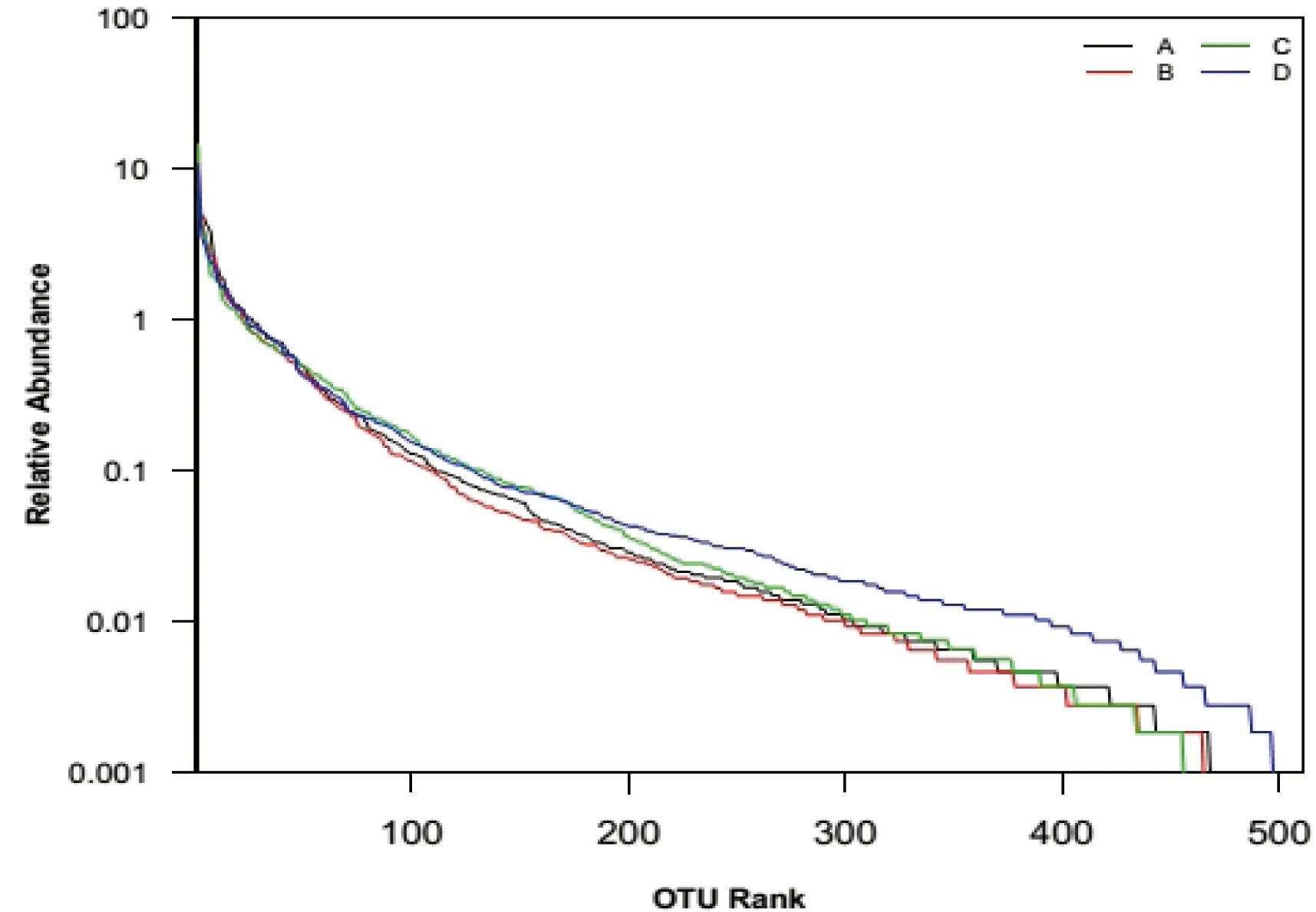
图4 高通量测序分析沙湖水体细菌多样性
2.4 主成分分析
通过PCA分析不同样本OTU(97%相似性)组成来反映样本间的差异[8]。本研究中PC1贡献度为65.66%,PC2贡献度为12.24%,累计贡献率高达77.9%,是变异的主要来源,可以解释变量的绝大部分信息。如图5所示,样品A1、A2、B1和B2中的细菌群落结构相似;样品C3和样品D3距离最近,说明两个取样点间细菌群落结构最为相似;样品C2和样品A1在PC2轴方向上间隔最大,说明两个取样点的细菌群落结构差异较大,且PC2是导致两个取样点差异的主要因素;A1和B3距离较大,说明两个取样点的细菌群落结构差异较大,且PC1是导致两个取样点差异的主要因素。
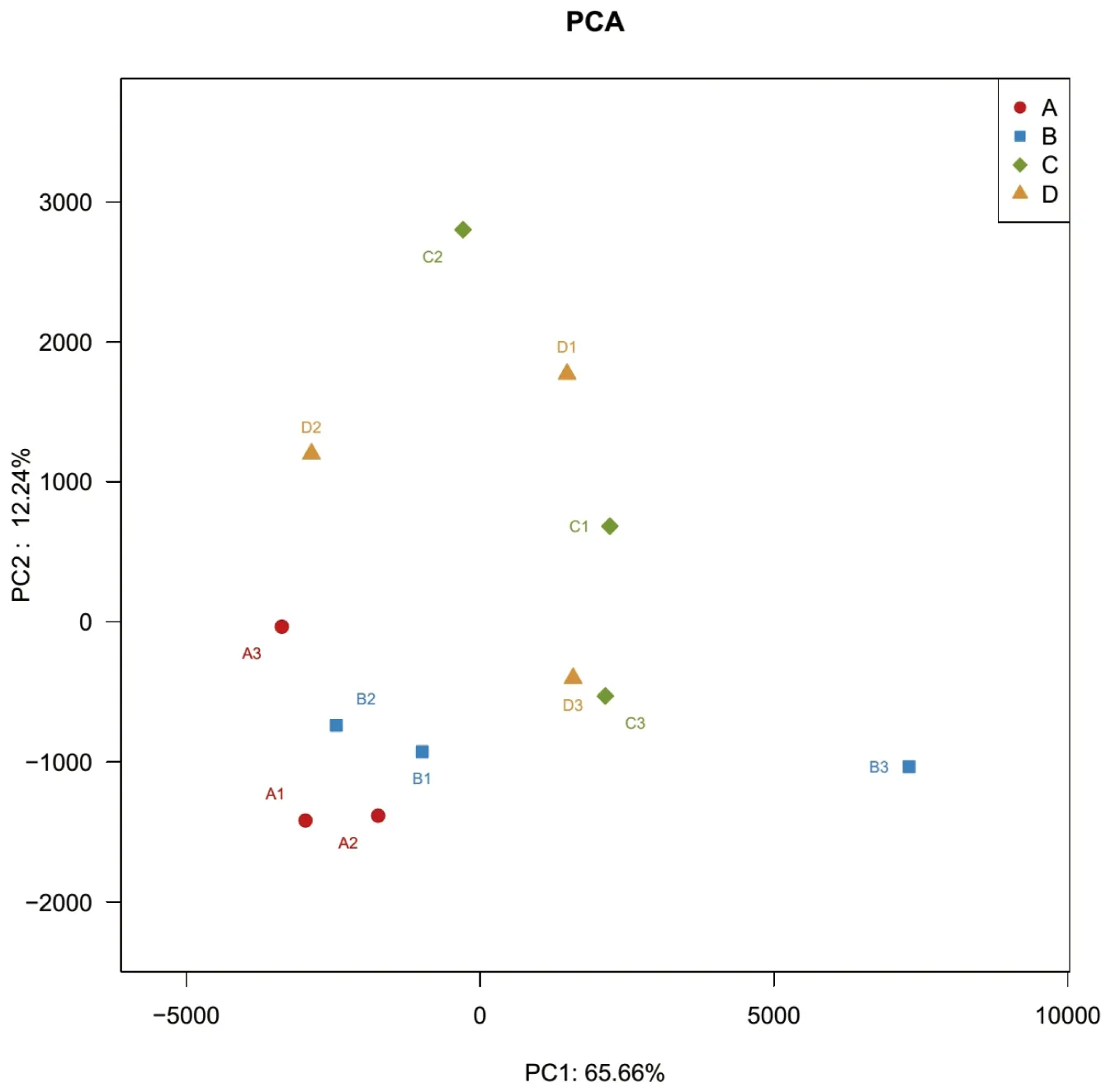
图5 主成分分析
3 讨论
细菌多样性是湖泊生态系统中食物链和食物网的重要组成部分,驱动着湖泊生态系统中绝大多数生物活性元素的生物地球化学循环[11]。本研究采用高通量测序技术对沙湖水体细菌多样性进行了分析,根据沙湖面积的大小及其形状以及邱小琮等[4]的报道,布设了12个采样点,共获得427 975条优化序列,覆盖度数据达99%以上,可以反映沙湖水体细菌群落组成的真实情况。优势类群主要是变形菌门、蓝细菌门、放线菌门和拟杆菌门等4大类群,占细菌总量的80%以上。这与任丽娟等[11]报道的在湖泊水体中主要以变形菌门、蓝细菌门、拟杆菌门、放线菌门和疣微菌门等5个淡水细菌门类基本一致,说明这些类群在湖泊水体生物地球化学循环中发挥着重要的作用。但在不同的取样位点,以上四大类群细菌相对丰度变化没有明显规律,这可能与湖泊面积大小以及水体流动造成营养物质、光照、氧气以及微生物分散程度不同等密切相关。这些因素会影响水体中微生物的多样性以及均匀度, 本研究中shannon指数范围在4.24~4.62之间,与浅水淡水湖泊——鄱阳湖类似[12](表层水体细菌多样性shannon指数为4.18),明显低于养殖水体[13](shannon指数范围在4.82~5.33),高于青藏高原东北部10个湖区细菌多样性[14](shannon指数范围在0.56~1.48),这也说明湖泊水体中微生物多样性的高低会受到不同区域、湖泊类型、富营养化程度、海拔高低等因素的影响,或者不同尺度条件下微生物多样性差异也会大不相同。在微生物群落结构组成上,与未受到严重污染的我国最大的淡水湖泊——鄱阳湖[12]相比,沙湖水体中除了优势类群变形菌门、拟杆菌门和放线菌门外,蓝细菌门出现且相对含量较高,这与当地工农业经济的发展,特别是农田灌溉退水以及水产养殖生产等使得过量营养盐进入沙湖,致使沙湖水体的富营养化所致[15]。本研究通过MiSeq测序技术对沙湖水体进行测序,首次全面揭示了宁夏沙湖水体内的细菌的丰度和多样性分析。这些结果将为沙湖水体质量的预测和理解沙湖水体中物质的生物地球化学循环提供理论基础。
[1]柴丽红, 崔晓巧, 彭 谦, 等.青海两盐湖细菌多样性研究[J]. 细菌学报, 2004, 44(3):271-275.
[2]RINKE C, SCHWIENTEK P, SCZYRBA A, et al. Insights into the phylogeny and coding potential of microbial dark matter[J]. Nature, 2013, 499(7459):431-437.
[3]崔安琪,翟 昊,于洪贤.宁夏沙湖浮游动物群落结构及多样性[J].东北林业大学学报,2015,43(9):121-124.
[4]邱小琮,赵红雪.宁夏沙湖浮游植物群落结构及多样性研究[J]. 水生态学杂志, 2011,32(1):20-26.
[5]朱明莹,于洪贤,马成学,等. 沙湖浮游植物多样性分析及水质评价[J].水产学杂志,2015,28(3):39-43.
[6]WANG P, CHEN B, YUAN R, et al. Characteristics of aquatic bacterial community and the influencing factors in an urban river[J]. Science of the Total Environment, 2016, 569-570:382-389.
[7]SCHLOSS P D, GEVERS D, WESTCOTT S L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies[J]. PLoS One, 2011, 6(12): e27310.
[8]WANG Y, SHENG H F, HE Y, et al. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags[J]. Applied Environmental Microbiology, 2012, 78(23):8264-8271.
[9]SRINIVASAN S, HOFFMAN N G, MORGAN M T, et al. Bacterial communities in women with bacterial vaginosis: high resolution phylogenetic analyses reveal relationships of microbiota to clinical criteria[J]. PLoS One, 2012, 7(6): e37818.
[10]BATES S T, CLEMENTE J C, FLORES G E, et al. Global biogeography of highly diverse protistan communities in soil[J]. The ISME Journal, 2012, 7(3):652-659.
[11]任丽娟, 何 聃, 邢 鹏, 等. 湖泊水体细菌多样性及其生态功能研究进展[J]. 生物多样性, 2013, 21(4): 421-432.
[12]寇文伯, 黄正云, 张 杰, 等. 鄱阳湖湖泊细菌群落组成及结构—以松门山为例[J]. 生态学报,2015, 35(23): 7608-7614.
[13]侯婷婷, 钟志平, 刘 缨, 等. 青石斑鱼海水循环水养殖水体的细菌群落特征[J]. 微生物学报, 2016, 56(2): 253-263.
[14]李 明, 郭 嘉, 石正国, 等. 春季青藏高原东北部湖泊细菌种类组成[J]. 应用与环境生物学报, 2013, 19(5):750-758.
[15]邱小琮, 赵红雪, 孙晓雪. 宁夏沙湖浮游植物与水环境因子关系的研究[J]. 环境科学, 2012, 33(7):2265-2271.
BacterialdiversityanalysisinShahuLakebased-onhigh-throughputsequencing
ZHANG Yan-ling1, LI Jing-yu1, LIU Jian-li1, LIU Ya-qin1, ZHANG Yi-fei2, ZHAO Ji3, DENG Wei1
(1. College of Biological Science & Engineering, Beifang Univesity of Nationality, Yinchuan 750021; 2. Inner Mongolia Academy of Environmental Sciences, Hohhot 010010; 3. College of Environment & Resources, Inner Mongolia University, Hohhot 010021, China)
In order to investigate the structure and diversity of microbial community in Shahu Lake, the total DNA of bacteria from water samples was extracted by E.Z.N.A.®Water DNA extraction kit, then the V4-V5 region of 16S rRNA gene was sequenced by MiSeq technology. Mothur software and R language were used to analyze the microbial community structure and diversity. A total of 427975 optimized sequences were obtained, and 20 Phylum, 50 Class, 95 Order, 161 Family, and 276 Genus were predicted at the 97% similarity level. The OTUs for A, B, C and D were 497, 496, 487 and 511, respectively. Proteobacteria, Cyanobacteria, Actinobacteria and Bacteroidetes are dominant groups. The microbial community structure in Shahu Lake is complex, but the composition and diversity of microbial community structure are not significant at different sites.
microbial communities; Shahu Lake; water; MiSeq sequencing
2016-11-15;
2016-11-21
国家自然科学基金(41661053);宁夏自然科学基金(NZ15098);北方民族大学引进人才科研启动项目(44/4400302502)
张燕灵, 专业方向为生物工程, E-mail:2806186414@qq.com
李靖宇,博士,讲师,研究方向环境微生物学、微生物生态学, E-mail: lijingyu1986@126.com
10.3969/j.issn.2095-1736.2017.06.056
Q93
A
2095-1736(2017)06-0056-04
