(S)-(+)-2,2-二甲基环丙烷甲酰胺的不对称合成*
2016-01-17张华林姚建华任春梅夏明珠王风云南京理工大学工业化学研究所江苏南京0094南京化工职业技术学院江苏南京0048
张华林,姚建华,任春梅,雷 武,夏明珠,王风云(.南京理工大学工业化学研究所,江苏南京 0094; .南京化工职业技术学院,江苏南京 0048)
(S)-(+)-2,2-二甲基环丙烷甲酰胺的不对称合成*
张华林1,姚建华1,任春梅2,雷武1,夏明珠1,王风云1
(1.南京理工大学工业化学研究所,江苏南京210094; 2.南京化工职业技术学院,江苏南京210048)
摘要:以环己二胺作为手性源,与3,5-二叔丁基水杨醛经缩合反应制得Salen配体(R,R)-N,N'-二(3,5-二叔丁基水杨醛)-1,2-环己二胺(6);甘氨酸乙酯盐酸盐经重氮化和6与三氟甲烷磺酸亚铜催化的不对称环丙烷化反应制得(S)-(+)-2,2-二甲基环丙烷甲酸乙酯(9); 9经水解、酰氯化和氨解反应合成了(S)-(+)-2,2-二甲基环丙烷甲酰胺,ee值82%,总收率55.4%,其结构经1H NMR和IR确证。
关键词:Salen配体;重氮乙酸乙酯;环丙烷化;(S)-(+)-2,2-二甲基环丙烷甲酸乙酯;(S)-(+)-2,2-二甲基环丙烷甲酰胺;不对称合成
泰能[1]是西司他丁与碳青霉烯类抗生素亚胺培南按1∶1制得的复合制剂。泰能的抗菌活性强,抗菌谱广,是目前临床上用于治疗重症感染的首选药物之一[2-3]。(S)-(+)-2,2-二甲基环丙烷甲酰胺(1)是合成西司他丁的关键中间体,因此1的合成具有重要意义[4]。

Scheme 1
目前1的合成方法主要有生物合成法和化学合成法。生物合成法需要特定的水解酶,且原料难以获得,在一定程度上限制了其发展[5-6]。化学合成法又分为不对称合成法和外消旋体拆分法。拆分法是国内工业中的生产方法,较长的合成路线和较低的拆分收率是拆分法的主要缺点。因此,不对称合成法成为研究关注的重点。Mori等[7]用异丁烯醛与(2R,4R)-2,4-戊二醇形成缩醛,与二碘甲烷进行不对称环丙烷化反应,臭氧作用下水解获得1,光学纯度94%。该反应条件苛刻、工艺复杂,难以工业化生产; Fujisawa等[8]在手性聚合奎尼定的催化下,乙烯醛与三氯乙醛环合加成制得(R)-β-三氯甲基-β-丙醇酸内酯,再经5步反应合成1。但乙烯醛不稳定,容易氧化,反应工艺复杂,总收率不高; Wang[9]以手性金属卡宾与异丁烯进行不对称环丙烷化反应,经氧化制得1。该方法反应条件苛刻,工业生产中难以实现。目前,以异丁烯为原料,在手性催化剂作用下与重氮乙酸酯反应合成1的方法最具工业化价值,所使用手性催化剂主要有以下几种:席夫碱类配体、半咕啉类配体、双噁唑啉类配体、其它C2轴手性配体等[10-11]。
本文参考文献[12-16]方法,以L-(+)-酒石酸(2)作为拆分试剂拆分1,2-环己二胺(3)制得(R,R)-1,2-环己二胺-L-(+)-酒石酸盐(4); 4 与3,5-二叔丁基水杨醛(5)经缩合反应制得Salen配体(R,R)-N,N'-二(3,5-二叔丁基水杨醛)-1,2-环己二胺(6),6中3具有刚性结构,能保持其构型,5的两个大取代基能够提高催化剂的对映体选择性。甘氨酸乙酯盐酸盐(7)经重氮化反应制得重氮乙酸乙酯(8); 8与异丁烯在6与三氟甲烷磺酸亚铜催化下经不对称环丙烷化反应制得(S)-(+)-2,2-二甲基环丙烷甲酸乙酯(9); 9经氢氧化钠溶液水解制得(S)-(+)-2,2-二甲基环丙烷甲酸(10); 10与固体光气(BTC)反应制得(S)-(+)-2,2-二甲基环丙烷甲酰氯(11); 11经氨解反应合成了1(Scheme 1),总收率55.4%,ee值82%,其结构经1H NMR和IR确证。
1 实验部分
1.1仪器与试剂
X-5型显微熔点仪(温度未校正); WZZ-2S型数字自动旋光仪; Bruker Avance(Ⅲ)300MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标); TENSOR-27型傅里叶快速红外光谱仪(KBr压片); SP6890型气相色谱仪;岛津LC-10ATVP型液相色谱仪; MAT-8200型质谱仪。
4[17-18]和7[19]按文献方法合成;其余所用试剂均为化学纯,南京化学试剂有限公司。
1.2合成
(1)6的合成
在三口瓶中依次加入4 1.49 g(5.64 mmol),碳酸钾1.60 g(11.58 mmol)和蒸馏水10 mL,搅拌使其溶解;加入乙醇38 mL,升温至回流,缓慢滴加5 2.70 g(11.52 mmol)的乙醇(12.7 mL)溶液,滴毕(用少许乙醇洗涤恒压滴液漏斗);反应3 h。加入蒸馏水10 mL,反应2 h;降温至5℃,保温2 h。抽滤,滤饼用二氯甲烷22.6 mL溶解,依次用水和饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏,残余物用无水乙醇重结晶得黄色粉末6 2.9 g,收率94%,纯度99%,m.p.211℃~212℃,[α]2D0-321°(c 1,CHCl3,下同)(m.p.210℃~211℃,[α]2D0-322°[18])。
(2)8的合成
在四口烧瓶中依次加入7 20.00 g(0.143 mol),水36 mL和二氯甲烷85 mL,搅拌使其溶解;冷却至-10℃,分6次加入亚硝酸钠12.00 g(0.174 mol)的冰水(36 mL)溶液,缓慢滴加5%硫酸13 mL,滴毕;反应0.5 h。静置分层,水层用少量冷二氯甲烷多次萃取,合并有机相,依次用冷5%碳酸氢钠溶液洗涤至中性,无水硫酸钠干燥,于室温减压蒸除溶剂得黄色油状液体8 13.9 g,收率85%。
(3)9的合成
在四口烧瓶中依次加入二氯甲烷226 mL,6 0.66 g(1.2 mmol),三氟甲基磺酸亚铜0.26 g(1.2 mmol)和8 0.23 g(2 mmol),回流反应0.5 h;通入异丁烯气体,同时缓慢滴加8 13.70 g(120 mmol)的二氯甲烷30 mL溶液,滴毕;反应16 h。蒸馏回收大部分二氯甲烷,过滤,滤液依次用饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏得无色透明液体9 12.80 g,收率64%(以7计),纯度98.5%;1H NMR δ:4.03(m,2H,OCH2),1.49(dd,J=8.0 Hz,5.5 Hz,1H,CH),1.16(m,3H,CH3in Et),1.11(d,J=5.3 Hz,6H,CH3CCH3),0.99(m,1H,CH2),0.85(dd,J=8.0 Hz,4.0 Hz,1H,CH2); IR ν:2 950(C-H),1 724(C=O),1 168(C-O)cm-1。
(4)10的合成
在三口烧瓶中依次加入9 7.00 g(49 mmol)和10%氢氧化钠溶液27 mL,搅拌下于80℃反应4 h。冷却至室温,用少量二氯甲烷洗涤去除未反应的有机杂质,用稀盐酸调至pH 1,用二氯甲烷(2×8 mL)萃取,合并萃取液,依次用饱和食盐水洗涤至中性,无水硫酸镁干燥,减压蒸除溶剂得无色透明液体10 5.3 g,收率94.3%,纯度99%,[α]20D115°([α]20D140.1°[20]),ee 82%;1H NMR(CDCl3)δ:11.49(s,1H,CO2H),1.50(m,1H,CH),1.26(s,3H,CH3),1.18(s,3H,CH3),1.13(m,1H,CH2),0.92(m,1H,CH2); IR ν:2 955(O-H),1 694(C=O),1 428,968(O-H),1 378(C-H),1 220,1 120(C-O)cm-1。
(5)1的合成
在三口烧瓶中依次加入10 10.00 g(88 mmol),二氯乙烷38 mL和BTC 8.90 g(0.03 mol),搅拌下滴加少量DMF,于70℃反应4 h。减压回收溶剂,残余物于60℃/6 KPa条件下保持1 h去除BTC,反应液加至装有二氯甲烷(100 mL)的四口烧瓶中,通入氨气,反应3 h。过滤,滤液蒸干后用乙酸乙酯(23 mL)溶解待用,滤饼真空干燥后于50℃用乙酸乙酯(110 mL)洗涤1 h,过滤(除去盐),合并乙酸乙酯相,于0℃析晶2 h,过滤得白色片状晶体,滤液蒸除大部分溶剂后冷却析晶,合并两次产品得1 9.0 g,收率90.7%,纯度99%,m.p.135℃~137℃,[α]2D085°(m.p.136℃~137.5℃,[α]2D0101.4°[2]);1H NMR δ:7.39(s,1H,NH2),6.70(s,1H,NH2),1.39(m,1H,CH),1.09(d,J=3.9 Hz,6H,CH3),0.82(m,1H,CH2),0.62(m,1H,CH2); IR ν:3 339,3 166,1 658,1 625(N-H),1 432(C-N)cm-1。
2 结果与讨论
2.1合成
在3的拆分过程中,温度的控制对产品光学活性影响较大。反应温度过高,对映选择性下降;而温度过低,体系粘稠,难以搅拌均匀,收率下降。
在6的合成中,为了使4的环上两个氨基均变成双亚胺基团,5应稍过量。5有两个大的给电子基团叔丁基,使其与4反应非常容易,且生成的6难溶于乙醇中,产物纯度较高,再经无水乙醇重结晶后纯度大于99%。
在8的合成中,加料顺序对收率影响较大。我们还尝试了先加入7的水和二氯甲烷混合溶液,用5%硫酸调至pH 1,然后将该溶液缓慢滴加至亚硝酸钠的水溶液中,反应完毕后处理同方法1.2(2),得黄色油状液体8 12.3 g,收率75%。两种方法相比,方法1.2(2)较优。
在1的合成中,还尝试了将11溶液缓慢滴加至-5℃的浓氨水(70 g)中,反应结束后。过滤,用冷水洗涤滤饼,滤饼烘干后经乙酸乙酯重结晶得1 8.0 g,收率80.7%,纯度99%。比较两种合成路线可知,方法1.2(5)收率较高,原因可能是将11滴加至冰冷的浓氨水中,反应放热剧烈,同时非常活泼的11有一小部分与水发生水解反应,生成了羧酸,从而导致产品收率降低。
2.2 9的合成工艺优化
为了寻找9的最佳合成工艺,分别考察了溶剂用量和催化剂用量对反应的影响,结果见图1~图3。
由图1可见,由于异丁烯是气体,难以大量溶于二氯甲烷中,为了提高8的转化率,必须加大溶剂用量以溶解较多的异丁烯,从而提高反应收率。实验表明,当溶剂用量增加至226 mL时,收率最高(64%);继续增大溶剂用量对收率影响不大。所以溶剂用量选择226 mL为宜。

图1 溶剂用量对9收率的影响Figure 1 Effect of solvent amount on the yield of 9

图2 催化剂用量对9 ee值的影响Figure 2 Effect of catalyst amount on ee value of 9
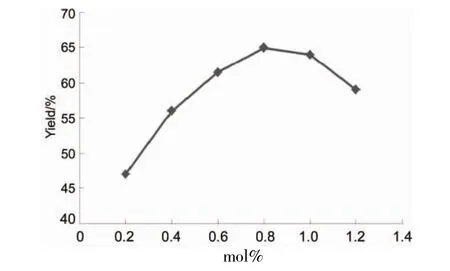
图3 催化剂用量对9收率的影响Figure 3 Effect of catalyst amount on the yield of 9
由图2可见,增加催化剂用量能够提高9的光学纯度。催化剂用量为1.0 mol%时9 ee值最高(82%);继续增加催化剂用量,ee值变化不大,同时收率明显降低。
由图3可以看出,催化剂用量为0.8 mol%时,收率最高(64%),减少或增加催化剂用量均会降低收率。这可能是因为催化剂量过多时,Cu(Ⅰ)络合物与8反应生成的铜-卡宾中间体未完全与异丁烯反应,分子间反应消耗了部分8,从而导致收率降低。综合考虑反应收率与光学纯度因素,选择催化剂用量为1.0 mol%较佳。
2.3 10的合成工艺优化
在10的合成中,考察了水解温度和反应时间对收率的影响,结果分别见图4和图5。由图4可见,水解温度为80℃时,收率最高(94.3%)。反应时间为4 h时,收率最高(95%),延长反应时间收率提高不明显。综合考虑,合成10的最佳反应条件为:于80℃反应4 h。
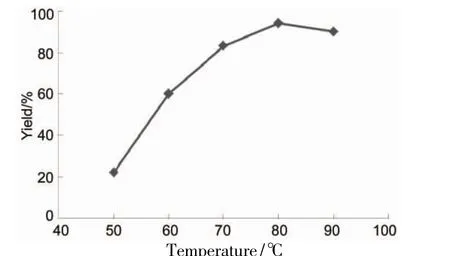
图4 水解温度对10收率的影响Figure 4 Effect of hydrolysis temperatureon the yield of 10

图5 反应时间对10收率的影响Figure 5 Effect of reaction time on the yield of 10
3 结论
合成了Salen配体(R,R)-N,N'-二(3,5-二叔丁基水杨醛)-1,2-环己二胺(6);以6和Cu(Ⅰ)形成的络合物为催化剂,甘氨酸乙酯盐酸盐为起始原料,经重氮化和不对称环丙烷化等反应合成了1,ee值82%,基本达到工业化拆分标准,收率55.4%,远远高于工业化生产水平。
该合成方法具有路线简单、反应条件温和、收率提高明显等优势,为1的合成研究提供了参考。
参考文献
[1]樊厚德.泰能的药理作用及临床应用概述[J].临床荟萃,2002,12(6):369-372.
[2]石晓华.西司他丁合成研究[D].浙江:浙江大学,2005.
[3]刘晓东,张铮,秦海东,等.泰能亚胺培南治疗重症肺炎40例临床分析[J].中国实用医药,2011(20):152-153.
[4]Stones G,Tripoll R,McDavid C L,et al.Investigation of macrocyclisation routes to 1,4,7-triazacyclononanes:Efficient synthesis from 1,2-ditosylamides[J].Organic and Biomolculer Chemistry,2008,6(1):374-384.
[5]Jin S J,Zheng R C,Zheng Y G,et al.R-enantioselective hydrolysis of 2,2-dimethylcyclopropanecarboxamide by amidase from a newly isolated strain Brevibacterium epidermidis ZJB-07021[J].Journal of Applied Microbiology,2008,105(4):1150-1157.
[6]Yang Z Y,Ni Y,Lu Z Y,et al.Industrial production of S-(+)-2,2-dimethylcyclopropanecarboxamide with a novel Recombinant R-amidase from Delftia tsuruhatensis [J].Process Biochemistry,2011,46(1):182-187.
[7]Mori A,Arai I,Yamamoto H.Asymmetric Simmons -Smith reactions using homochiral protecting groups [J].Tetrahedron,1986,42(23):6447-6458.
[8]Fujisawa T.Regioselective ring cleavage of chiral-βtrichloromethyl-β-propiolactone with organoaluminum compounds for the synthesis of optically active intermediates[J].Tetrahedorn Letters,1998,39(52):9735-9738.
[9]Wang Q W,Wang F K.The synthesis of S-(+)-2,2-dimethylcyclopropane carboxylic acid:A precursor of cilastatin[J].Tetrahedron:Asymmetry,1998,9(22):3971-3977.
[10]高金山,边庆花,郭洪超,等.手性金属配合物催化的不对称环丙烷化反应的新进展[J].有机化学,2007,27(4):438-448.
[11]Singh V K,Dattagupta A,Sekar G.Catalytic enantioselective cyclopropanation of olefins using carbenoid chemistry[J].Snythesis,1997,8(20):137-149.
[12]张治国,王新根,徐官根.一种S-(+)-2,2-二甲基环丙烷甲酰胺的合成方法[P].CN 101 735 099A,2010.
[13]姚小泉,陈惠麟,郑卓.烯烃不对称催化环丙烷化反应进展评述[J].化学进展,2000,12(3):282-294.
[14]Paquette L A.Chiral Reagents for Asymmetric Synthesis:不对称合成中的手性试剂[M].侯雪龙,吴劼.译.华东理工大学出版社,2006:202-207.
[15]武立鲲,张福利.(S)-(+)-2,2-二甲基环丙烷甲酰胺的合成[J].中国医药工业杂志,2011,42(11):491-494.
[16]Kim J S,Son J M,Roh K R,et al.Process for preparing optically active cyclopropane carboxamide and derivatives thereof[P].200 706 9841,2007.
[17]Larrow J F,Jacobsen E N,Gao Y,et al.A practical process for the large-scale preparation of(R,R)-N,N'-bis(3,5-di-tert-butyl-salicylidene)-1,2-cyclohexanediaminomanganese(Ⅲ)chloride,a highly enantioselective epoxidation catalyst[J].Journal of Organic Chemistry,1994,59:1939-1940.
[18]梁斌.CO2与环氧丙烷不对称环加成反应催化体系的设计与研究[D].大连:大连理工大学,2004.
[19]门宝琴.氨基酸酯及其衍生物的合成[D].南京:东南大学,2005.
[20]Thomas Meul.Process for preparation of optically active carboxylic acids[P].CH 682 485,1993.
·研究简报·
Asymmetric Synthesis of
(S)-(+)-2,2-dimethylcyclopropane Carboxamide
ZHANG Hua-lin1,YAO Jian-hua1,REN Chun-mei2,LEI Wu1,XIA Ming-zhu1,WANG Feng-yun1
(1.School of Chemistry&Engineering,Nanjing University of Science&Technology,Nanjing 210094,China; 2.Nanjing College of Chemical Technology,Nanjing 210048,China)
Abstract:A Salen ligand(6)was prepared by condensation reaction of chiral diaminocyclohexane with 3,5-ditertbutyl-salicylaldehyde.(S)-(+)-2,2-dimethylcyclopropane carboxylic acid ethyl ester(9)was obtained by diazotization and asymmetric cyclopropanation catalyzed by 6 and copper(Ⅰ)triflate from glycine ethyl ester hydrochloride.(S)-(+)-2,2-dimethylcyclopropane carboxamide with an overall yield of 55.4% and 82% ee was synthesized eventually by hydrolysis,chlorization and ammonolysis from 9.The structure was confirmed by1H NMR and IR.
Keywords:Salen ligand; ethyl diazoacetate; cyclopropanation;(S)-(+)-2,2-dimethylcyclopropane carboxylic acid ethyl ester;(S)-(+)-2,2-dimethylcyclopropane carboxamide; asymmetric synthesis
作者简介:张华林(1990-),男,汉族,江苏淮安人,硕士研究生,主要从事有机合成研究。E-mail:zhang37169947@126.com
收稿日期:2014-10-14
DOI:10.15952/j.cnki.cjsc.1005-1511.2015.05.0423 *
文献标识码:A
中图分类号:O621.3; O624.5
通信联系人:任春梅,讲师,E-mail:346728312@ qq.com
