卡培他滨关键中间体精制工艺改进
2015-12-13李孝常臧恒昌
代 炎,李孝常,臧恒昌,裴 林
(1.山东大学药学院,山东济南 250012;2.合肥立方制药股份有限公司,安徽 合肥 230088)
卡培他滨(kapecitabine,化合物4),化学名为5'-脱氧- 5-氟-N-[(戊氧基)羰基]胞苷[1-5],由瑞士Hoffmann-La Roche公司研发的一种新型5-氟尿嘧啶(5-FU)前体药物,1998年首次在瑞士上市。本品是一种新型口服氟胞嘧啶核苷类似物,本身无细胞毒性,在体内酶的作用下代谢为5-氟尿嘧啶(5-FU),进而发挥抗肿瘤作用,临床主要用于治疗对紫杉醇和多柔比星等药物无效的晚期原发性或转移性乳腺癌、结/直肠癌及其它实体瘤[6-14]。奥沙利铂联合表阿霉素与卡培他滨一线治疗晚期胃癌[15],吉西他滨联合卡培他滨治疗复发转移性乳腺癌[16]。
卡培他滨的化学合成有多条途径[17-22]。其中,以5'-脱氧-5-氟尿苷(化合物1)为原料的合成工艺,由于所用的保护基试剂价廉,工艺条件易于控制等优点而被广泛采用。它的合成路线如图1。
化合物3是合成卡培他滨的一个关键中间体(下称中间体2),其纯度之高低将直接影响后续合成和分离。根据陈越磊等[23]报道的中间体2的合成工艺条件为中间体1(化合物2)的氯化、胺化反应结束后,先减压蒸出溶剂,残余物在乙酸乙酯和水中分配,分出水层后有机相依次用稀盐酸、饱和碳酸氢钠溶液和饱和食盐水溶液洗涤,经干燥剂干燥后蒸出大部分溶剂,冷却后析出白色固体。该固体再用乙酸乙酯-石油醚重结晶精制得到中间体2,3-二苯甲酰基-5-脱氧-5-氟胞苷白色片状晶体,收率66%。
我们的仿制实验结果表明[24]:依上述方法所得到的固体粗品在乙酸乙酯—石油醚混合溶剂中重结晶效果欠佳。欲获得合格纯度的中间体2,必须经过2次以上重结晶过程。在工业生产上表现为精制周期长,溶媒耗损高,尤其是每次精制过程中的收率折扣,使得精制成本步步攀升。为了解决中间体2精制周期长,溶媒损耗高等问题,我们开展了中间体2精制方法的研究工作。
1 试验材料及仪器
1.1 试剂 2,3-二苯甲酰基-5-脱氧-5-氟尿苷,二甲胺基吡啶、乙腈、吡啶、三氯氧磷、浓氨水、乙酸乙酯、盐酸、碳酸氢钠、氯化钠、石油醚(60~90℃)均为分析纯;水为二次蒸馏(合肥立方制药股份有限公司自制);检测用乙酸、乙腈、甲醇均为色谱级。
1.2 仪器 Shimadzu LC-20ATvp高效液相色谱仪(岛津公司)、LC Solution色谱工作站(岛津公司)、电子分析天平(梅特勒-托利多)、超声波清洗器(天津奥特赛恩斯有限公司)。
1.3 检测方法[25]色谱柱:Kromasil C18(4.6 mm×260 mm,5μm);柱箱温度:40℃;自动进样室:5℃;流速:1 mL·min-1;进样量:10μL;检测器:D2 lamp;稀释剂为甲醇∶乙腈∶水 =7∶1∶12;样品浓度:0.6 mg·L-1;流动相:A 为0.1%醋酸,B 为甲醇∶乙腈∶A=7∶0.2∶12.8,C 为甲醇∶乙腈∶A=16∶1∶3;梯度洗脱:0→5 min;C在流动相中的比例(f)保持在0%;5→20 min,f由 0%上升至 51%;20→30 min,f保持在51%,30→31 min;f由51%下降至0,31→40 min,f保持在0;检测波长:250 nm;面积归一法计算纯度。
2 实验方法
2.1 中间体2的制备 将二甲氨基吡啶 (44.5 g,0.363 mol)悬浮于乙腈(280 mL)和吡啶(29.5 mL,0.364 mol)中,于 8℃条件下滴加三氯氧磷(33.7 g,0.22 mol),加毕,于23~25℃继续搅拌反应65 min后降至2℃。降温结束后,缓缓滴加2,3-二苯甲酰基-5-脱氧-5-氟尿苷(化合物2)(33.07 g,折纯0.073 mol)和乙腈(187 mL)制成的溶液。滴加完毕,升温至23~25℃,并继续搅拌反应3.5 h,再降温至5~7℃,并向此溶液中滴加浓氨水(176 mL),于23~25℃搅拌反应约70 min并以TLC法指示反应终点(展开剂:乙酸乙酯/石油醚 =7.5/2.5;固定相:GF254)。
2.2 提取、洗涤 反应结束后,于热水浴中减压浓缩回收溶媒至净,得到橙黄色半固状浓缩物。然后用乙酸乙酯(450 mL)和蒸馏水(350 mL)超声萃取,静置15 min后分出水层。有机相先后用2 mol·L-1的稀盐酸(165 mL),饱和碳酸钠溶液(150 mL)各洗涤一次。最后用饱和食盐水洗涤三次(200 mL+140 mL+100 mL)。分净水层后将无水硫酸钠(50 g)加入其中,振摇片刻后静止过夜。滤除干燥剂后得到干燥的黄色澄明的乙酸乙酯萃取液378 mL,再加乙酸乙酯至一定浓度。
2.3 沉淀除杂 将上述干燥的乙酸乙酯萃取液转入三角瓶中,置于冷藏室缓缓降温至适宜温度,静置一定时间后取出,并作真空抽滤,得到含各种杂质的淡黄色固体沉淀物。
2.4 结晶 将滤除杂质后的母液真空浓缩至一定体积时趁热加入活性炭(0.75 g)并置于沸水浴上回流脱色10 min后过滤除炭。将脱色液加乙酸乙酯稀释至一定浓度,将此溶液置于冷冻室缓缓降温至一定温度,并于同温下静置结晶一定时间后真空吸滤,并用少量相同冷溶媒洗涤2次。滤饼于60℃真空干燥至恒重,观察结晶物外观,检测纯度(液相方法),计算收率。
2.5 回收二析物 将一步结晶母液真空浓缩至约原体积的1/2时加入适量活性炭,并于水浴上回流脱色10 min,将滤液置于-2~0℃条件下冷却结晶12 h,再真空过滤,滤饼用冰冷的乙酸乙酯洗涤3次后依法干燥至干,得到白色二析物结晶。
3 实验结果与讨论
3.1 考察乙酸乙酯提取液的浓度对中间体2收率和纯度的影响 按“2.1”的方法(放大3倍)制备中间体2的乙酸乙酯提取液,滤除干燥剂后得到干燥的黄色澄明的乙酸乙酯萃取液1 150 mL,取300 mL萃取液,平均分成6份,分别加入规定量的乙酸乙酯稀释至不同的浓度,于0~3℃析晶,陈化12 h后,滤除杂质,母液经浓缩、脱色后浓度调节至85~90 g·L-1,于-6 ~-3℃析晶,陈化 10 h 后,过滤,滤饼于60℃真空干燥至恒重,考察中间体2的纯度和收率。结果见表1。
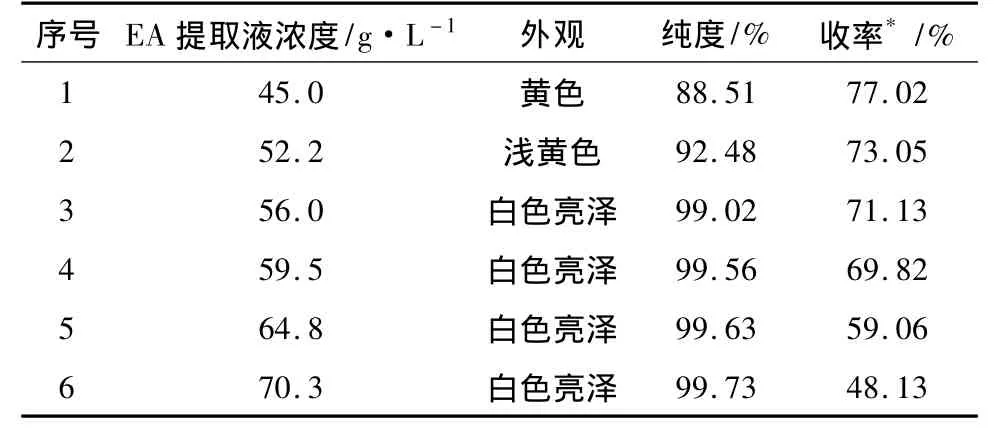
表1 乙酸乙酯提取液的浓度对中间体2收率和纯度的影响
由表1可知,随着中间体2乙酸乙酯提取液的浓度增大,纯度逐渐升高,当浓度增大到 56.0 g·L-1时,纯度达到99.02%,已经满足中间体2的质量要求(≥99%),再继续增加浓缩,虽然纯度还会提高,但是在沉淀去除杂质的同时,产品损失也增大,当浓度增大到 70.3 g·L-1时,收率只有48.13%。根据考察结果,提取液的浓度选择56~60 g·L-1。
3.2 考察杂质沉淀温度对中间体2收率和纯度的影响 取150 mL萃取液,加乙酸乙酯稀释至中间体2含量59.5 g·L-1,平均分成3份,于不同温度下进行沉淀,陈化12 h后,滤除杂质,母液经浓缩、脱色后浓度调节至 85~90 g·L-1,于-6~-3℃析晶,陈化10 h后,过滤,滤饼于60℃真空干燥至恒重,考察中间体2的纯度和收率。结果见表2。

表2 杂质沉淀温度对中间体2收率和纯度的影响
由表2可知,随着杂质沉淀温度的提高,纯度逐渐升高,当沉淀温度降至-3~-1℃时,纯度达到99.89%,但是随着温度的降低,中间体2也大量析出,导致收率较低(50.02%),当沉淀温度升至3~4℃时,部分杂质无法析出,导致中间体2的纯度偏低(92.86%)。根据考察结果,杂质沉淀温度选择0~2℃。
3.3 考察杂质陈化时间对中间体2收率和纯度的影响 取300 mL萃取液,加乙酸乙酯稀释至中间体2 含量59.5 g·L-1,平均分成6 份,于0 ~2℃下进行沉淀,陈化一定时间后,滤除杂质,母液经浓缩、脱色后浓度调节至 85~90 g·L-1,于-6~-3℃析晶,陈化10 h后,过滤,滤饼于60℃真空干燥至恒重,考察中间体2的纯度和收率。结果见表3。
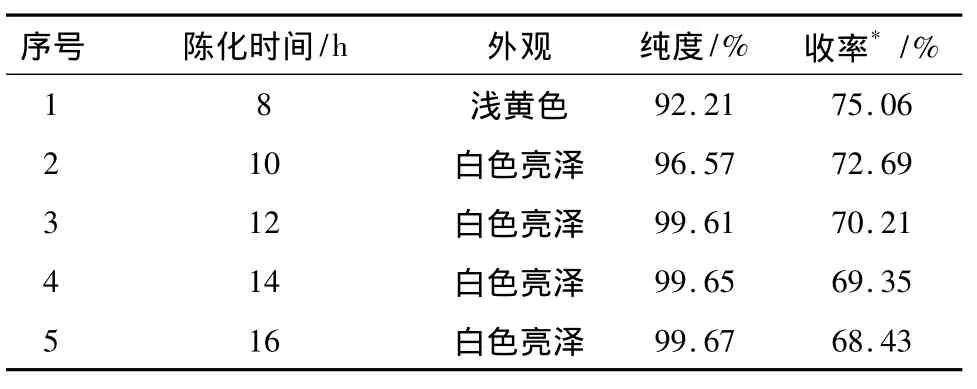
表3 杂质陈化时间对中间体2收率和纯度的影响
由表3可知,随着杂质陈化时间的延长,纯度逐渐变大,当陈化时间为8 h时,由于杂质未充分析出,导致中间体2纯度只有92.21%,当陈化时间延长至12 h时,中间体2纯度达到99.61%,再继续延长时间,纯度基本不变,为了提高效率,选择陈化时间为12~14 h。
3.4 考察中间体2析晶浓度对中间体2收率和纯度的影响 取300 mL萃取液,加乙酸乙酯稀释至中间体2含量59.0 g·L-1,平均分成 6份,于0 ~2℃下陈化12 h后,滤除杂质,母液经浓缩、脱色后浓度调节至一定浓度,于-6~-3℃析晶,陈化10 h后,过滤,滤饼于60℃真空干燥至恒重,考察中间体2的纯度和收率。结果见表4。
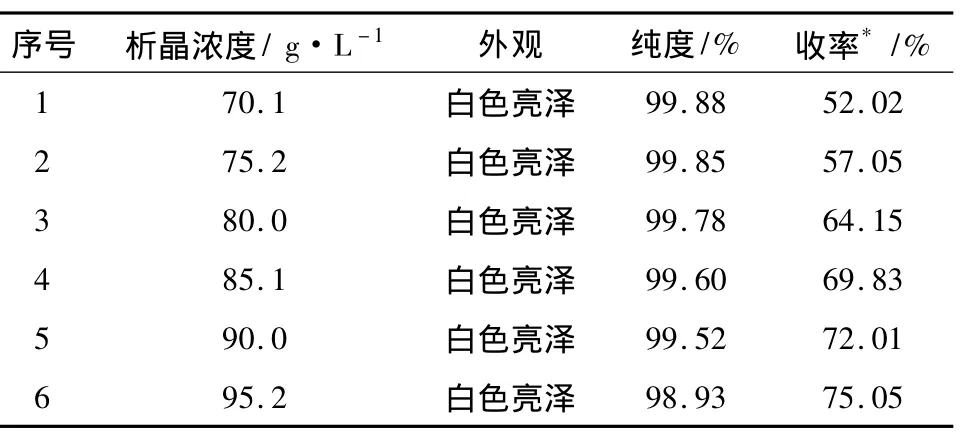
表4 析晶浓度对中间体2收率和纯度的影响
由表4可知,随着中间体2析晶的浓度增大,收率逐渐升高。当析晶浓度增大到85.1 g·L-1时,收率达到69.83%,纯度99.60%,当析晶浓度增大到95.2 g·L-1时,收率达到 75.05%,但纯度降低至98.93%,不满足中间体2的质量要求(≥99%)。析晶浓度越高,溶剂含量少,部分杂质在伴随着中间体2沉淀出来,导致纯度降低,根据考察结果,中间体2的析晶浓度选择85~90 g·L-1。
3.5 考察中间体2析晶温度对中间体2收率和纯度的影响 取200 mL萃取液,加乙酸乙酯稀释至中间体2含量58.3 g·L-1,平均分成4份,于0~2℃下陈化12 h后,滤除杂质,母液经浓缩、脱色后浓度调节至85~90 g·L-1,分别于不同温度进行析晶,陈化10 h后,过滤,滤饼于60℃真空干燥至恒重,考察中间体2的纯度和收率。结果见表5。
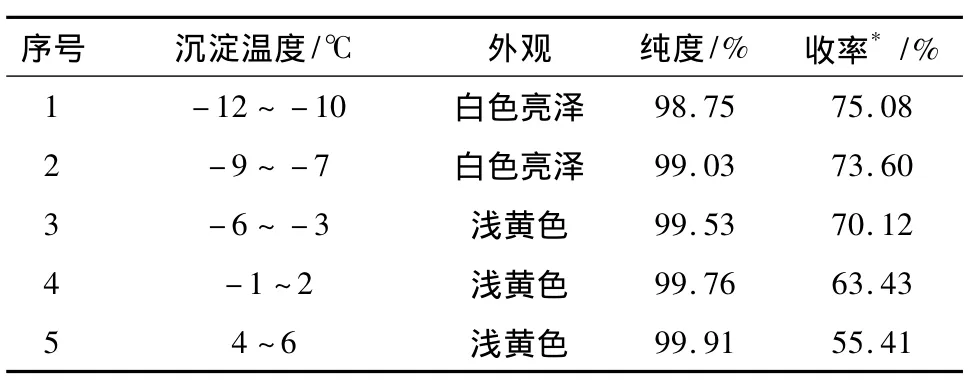
表5 析晶温度对中间体2收率和纯度的影响
由表5可知,随着析晶温度的升高,收率逐渐降低,纯度逐渐升高。当析晶温度为4~6℃时,纯度达到99.91%,但是收率较低,只有55.41%;当析晶温度的降低至-6~-3℃,收率升高至70.12%,当析晶温度的降低至-9~-7℃,收率升高至73.60%,纯度99.03%;在-12 ~-10℃时析晶,收率达到75.08%,但是纯度降至98.75%;析晶温度为-9~-3℃时,收率和纯度都符合要求,再继续降低温度,收率增加较小,并且产品纯度有所下降;从产品质量考虑,选择的析晶温度为-9~-3℃。
4 结论
经过我们创造性地研究,最终找到了一种将中间体2与其他所有未作用的原料、中间体1及合成杂质以一种最简便的方式获得近乎彻底地分离。先将洗净并干燥的乙酸乙酯提取液在规定的浓度、温度条件下静置适当时间,使所有杂质一次沉淀析出。滤除沉淀后将滤液浓缩至适宜的浓度、并在适宜的温度下直接结晶,从而直接获得优质目标产品。
该精制工艺过程简便、具有分离效率高、收率高、成本低廉的优势,尤其适合于工业化生产。
[1]李菊平,张博琛.卡培他滨合成工艺的研究[J].齐鲁药事,2012,31(8):442-443,447.
[2]党晓翠,赛达力木,赵 敏,等.卡培他滨的合成[J].中国新药杂志,2013,22(8):967-970.
[3]柴洪伟,尚惊鸿,王 维,等.卡培他滨的合成[J].中国医药工业杂志,2011,42(12):887-888,913.
[4]安富荣,戈升荣,祝德秋.卡培他滨的药理特性及临床应用进展[J].中国新药与临床杂志,2002,21(8):503-507.
[5]Shimma N,Umeda I,Arasaki M,et al.The design and synthesis of a new tumor-selective fluoropyrimidine carbamate,capecitabine[J].Bioorg Med Chem,2000,8(7):1697-1706.
[6]Miles D,Zielinski C,Martin M,et al.Combining capecitabine and bevacizumab in metastatic breast cancer:A comprehensive review[J].European Journal of Cancer,2012,48(4):482-491.
[7]潘战和,苏 安,王 馨,等.草酸铂联合卡培他滨一线化疗后卡培他滨维持治疗晚期胃癌[J].中国肿瘤临床,2012,39(20):1552-1555.
[8]安富荣,戈升荣,祝德秋.卡培他滨的药理特性及临床应用进展[J].中国新药与临床杂志,2002,21(8):503-507.
[9]张东伟,周晋华,吴飞雪.卡培他滨单药治疗老年晚期胃癌临床研究[J].肿瘤基础与临床,2008,21(3):238-239.
[10]余 靖,邸立军,宋国红,等.多西他赛联合塞替派与多西他赛联合卡培他滨治疗转移性乳腺癌的随机、对照临床研究[J].北京大学学报(医学版),2011,43(1):151-156.
[11]农先胜,黄显实.卡培他滨单药或联合用药治疗复发/转移性乳腺癌的疗效观察[J].中国癌症防治杂志,2012,4(4):336-339.
[12]李金燕.卡培他滨联合奥沙利铂与替吉奥联合奥沙利铂治疗进展期胃癌的效果比较[J].中国实用医药,2013,8(18):157-158.
[13]张 静,刘 俏,彭六保,等.以卡培他滨为基础的联合化疗方案治疗晚期胃癌新进展[J].中国药房,2013,24(12):1137-1140.
[14]周风举,彭 晔,王 宁,等.伊立替康联合卡培他滨一线治疗进展期胃癌临床观察[J].中华肿瘤防治杂志,2011,18(9):710-712.
[15]李云松,杜瀛瀛,卜丽佳,等.奥沙利铂联合表阿霉素与卡培他滨一线治疗晚期胃癌的临床观察[J].安徽医药,2014,18(12):2370-2372.
[16]蒋祥德,刘友如,施朕善.吉西他滨联合卡培他滨治疗复发转移性乳腺癌的临床观察[J].安徽医药,2014,18(2):335-336.
[17]何学军,肖亚东,王德才.卡培他滨合成路线图解[J].中国医药工业杂志,2009,40(7):549-551.
[18]陈莉莉,岑均达.卡培他滨的新合成路线[J].中国药物化学杂志,2010,20(4):275-277.
[19]赵明礼,赵玉涛,张 召,等.抗肿瘤药物卡培他滨的合成新方法[J].高等学校化学学报,2012,33(8):1733-1737.
[20]周振轩,蒋翔锐,吴玉林.卡培他滨的合成[J].化工时刊,2014,28(3):4-6.
[21]杨建楠,杜有国,宗在伟,等.卡培他滨的合成工艺研究[J].中国药师,2011,14(6):796-798.
[22]潘瑜群,陈国华,江传亮,等.卡培他滨的合成工艺改进[J].中国药物化学杂志,2011,21(5):376-378.
[23]陈越磊,岑均达.卡培他滨类似物的合成及体内抗肿瘤活性[J].中国药物化学杂志,2004,14(5):277- 279,282.
[24]季俊虬,高美华,李孝常,等.一种卡培他滨中间体的简便精制方法:中国,CN201310674404.4[P].2013-12-13.
[25]左家信,廖声华,严 拯.HPLC法测定卡培他滨的含量及有关物质[J].海峡药学,2013,25(1):48-51.
