先天性肌营养不良1A型颅脑磁共振成像和基因检测诊断分析:附两例报道及文献复习
2014-02-08张晓莉牛国辉杜开先徐发林贾天明
张晓莉,牛国辉,杜开先,徐发林,韩 瑞,贾天明
先天性肌营养不良(congenital muscular dystrophy,CMD)是一组异质性遗传性神经肌肉病,该疾病的分型有十余种,多以不同的致病基因为主,其中的先天性肌营养不良1A型(merosin-deficient congenital muscular dystrophy,MDC1A)与编码laminin-α2(又称merosin)的LAMA2基因突变有关,又称Merosin缺陷型,占总CMD的30%~40%,但该型在欧洲常见,我国报道很少,迄今国内共报道13例,仅3例有基因诊断[1-3]。现报道我院收治的2例MDC1A患儿,并结合国内报道的13例患儿做一总结,研究颅脑磁共振成像(MRI)和基因检测诊断的意义。
1 资料与方法
1.1 病例资料 先证者1,女,8月龄,因下肢无力于2013-07-16就诊。系第1胎第1产,足月顺生,无窒息。4月龄时发现扶站下肢不支撑,未行治疗,8月龄仍不能扶站,遂来我院就诊。查体:智能、语言正常,竖头稳,双手抓物灵活,可独坐,扶站下肢不支撑,膝腱反射消失,双下肢肌张力低,肌力4级,肌容积正常,上肢肌力正常。既往未曾治疗,家族史无特殊。查肌酸激酶(CK)2 500 U/L。肌电图示肌源性损害。颅脑MRI示脑白质营养不良(见图1)。诊断MDC1A。提取外周抗凝血4 ml,送康旭医学检验所通过二代测序法进行基因检测,并通过一代测序法进行验证,LAMA2基因检测发现复合杂合突变,一个是无义突变c.4048C>T(p.Arg1350*),一个是移码突变c.457-458insT(p.Ile153fs),均为杂合突变(见图2)。
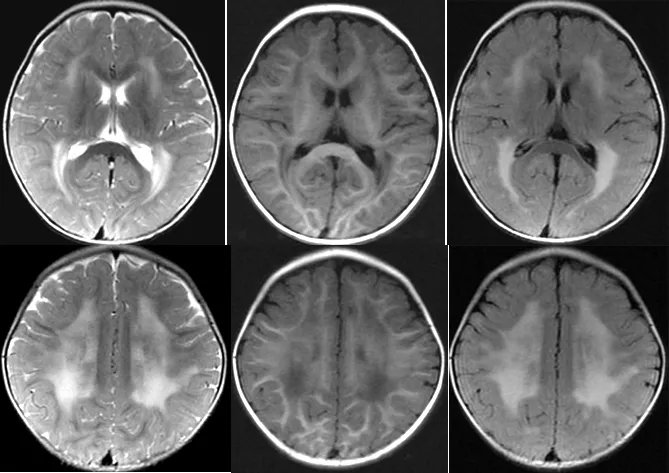
图1 先证者1颅脑MRI示脑白质广泛融合性脱髓鞘病变
Figure1 The brain MRI of proband 1 showing widely distributed fusion of white matter demyelination
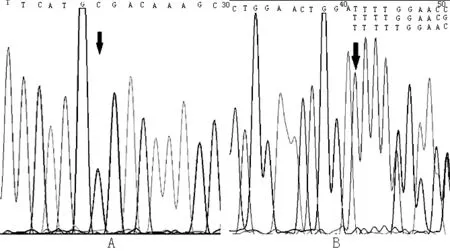
注:A图为无义突变,编码区第4048号碱基由C变为T,导致本该编码精氨酸的1350号密码子变为终止密码子;B图为移码突变,在编码区457号碱基和458号碱基之间插入了T,导致第153号密码子后发生移码变异
图2 先证者1 LAMA2基因测序图
Figure2 LAMA2-gene sequencing diagram of proband 1
先证者2,男,3岁4月,因发育落后,下肢无力,不会独站,遂于2013-10-25就诊。系第1胎第1产,足月剖宫产,出生体质量2 800 g,生后哭声稍低,无窒息抢救史。运动发育史:9个月时翻身,1岁会坐,现不会独站。查体:语言表达良好,智力正常,可独坐,不能站立,上肢肌力4级,双下肢肌力3级,双膝反射及跟腱反射消失,双足轻度内翻,无明显肌肉萎缩,四肢肌张力低下,以下肢明显。肌电图提示:神经肌肉混合性损害,CK 1 041 U/L,颅脑MRI示双侧脑室前角旁、体旁及后角旁对称性异常信号(见图3)。诊断MDC1A。
1.2 文献复习 通过万方数据库和中国知网中文数据库以“肌营养不良”和“先天性”为检索词在题名或关键词中检索2013年12月以前发表的中文学术期刊,并在PubMed数据库中查到中国人发表的先天性肌营养不良文献1篇,通过阅读全文,从中筛选出符合MDC1A的病例,共9 篇文献,13例患儿,结合本院的2例患儿,共15例,做一综合分析。
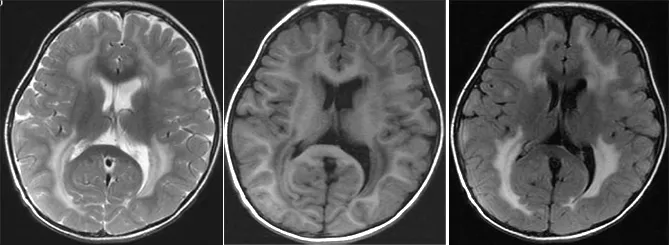
图3 先证者2颅脑MRI示侧脑室前角、体部、后角旁均可见融合性脱髓鞘病变
Figure3 The brain MRI of proband 2 showing the integration of demyelination in the front horn,the body and beside the posteior horn of lateral ventricle
2 结果
15例患儿中运动发育落后15例,CK升高14例,颅脑MRI弥漫性白质异常信号 14例,智力正常13例,腱反射消失13例(2例未描述),肌电图检查11例均显示肌源性损害,并周围神经损害2例,病理检查9例均为肌营养不良改变;基因检测4例,均发现LAMA2基因突变,其中2例为纯合突变,2例为复合杂合突变(见表1)。
3 讨论
MDC1A是最常见的CMD,欧美多见,我国罕见,目前报道共13例(其中临床诊断2例,病理诊断8例,基因诊断3例),其确诊目前主要依靠肌活检,但此检查方法多数医院不能进行。本研究通过对15例患儿共同特点的分析发现,颅脑MRI在诊断上具有特异性,而进一步基因检测可以确诊并可以指导产前诊断。
3.1 临床特点 回顾国内既往的病例报道和本文的2例共15例MDC1A患儿发现,所有病例在生后6个月内起病,生后早期即表现出明显的肌张力低下,运动发育迟滞,以端坐延迟(13/15)、不能扶站就诊者居多,最大获得运动功能多为独坐(13/15),仅2例获得行走能力,但为肌病步态。体格检查除发现肌张力低下、运动落后,均可见腱反射消失(13/13,2例未描述),此与中枢性疾病所导致的肌张力低下和发育落后不同。
血清学检查见CK水平明显升高,数倍至数十倍升高,但无临床心肌受损的表现。本组患儿中CK最高达9 640 U/L。本病患儿肌电图检查主要呈肌源性损害(11/11),部分同时存在周围神经损害(病例12和15),因为merosin蛋白除了存在于横纹肌细胞的基底膜外,在脑血管基底膜、发育中的白质纤维束均有表达,还存在于施旺细胞的基底膜,因此周围神经系统也可受累,表现为运动或感觉神经传导速度减慢[10],本文先证者2肌电图检查即发现胫后、腓总神经运动传导速度减慢,感觉传导未见异常。肌肉活检9例均呈肌营养不良改变,表现为肌纤维的坏死、再生,肌间纤维结缔组织增加,还有炎性细胞浸润,用特异性抗体行免疫组织化学染色显示merosin染色阴性,提示merosin蛋白缺失。merosin蛋白是层黏连蛋白-2的3个亚单位之一,参与细胞间的识别、细胞分化、塑形和迁移等,merosin缺乏可造成细胞骨架与细胞外基质的连接破坏,导致肌纤维变性、坏死。
3.2 颅脑MRI改变 MDC1A患儿多无明显的认知功能障碍(15例中仅2例智力稍差,余均正常),但常规行颅脑MRI检查均可见脑白质营养不良改变(15例患儿中有1例未描述颅脑MRI改变,其余14例均有该特异性改变),与常见的脑白质营养不良均有严重的认知功能障碍不同。这些脑白质病变的特点均为双侧对称,多为弥漫性白质异常,侧脑室旁白质和皮质下均可见到。既往认为merosin阴性患儿仅有脑白质的病变,近年来也有小脑、脑干发育不良的报道[11]。本文2例患儿颅脑MRI均表现为侧脑室前角、体部、后角旁广泛融合性脱髓鞘改变,未见小脑和脑干的异常。此种特征性的改变有助于本病的诊断,其病理学机制尚不清楚。
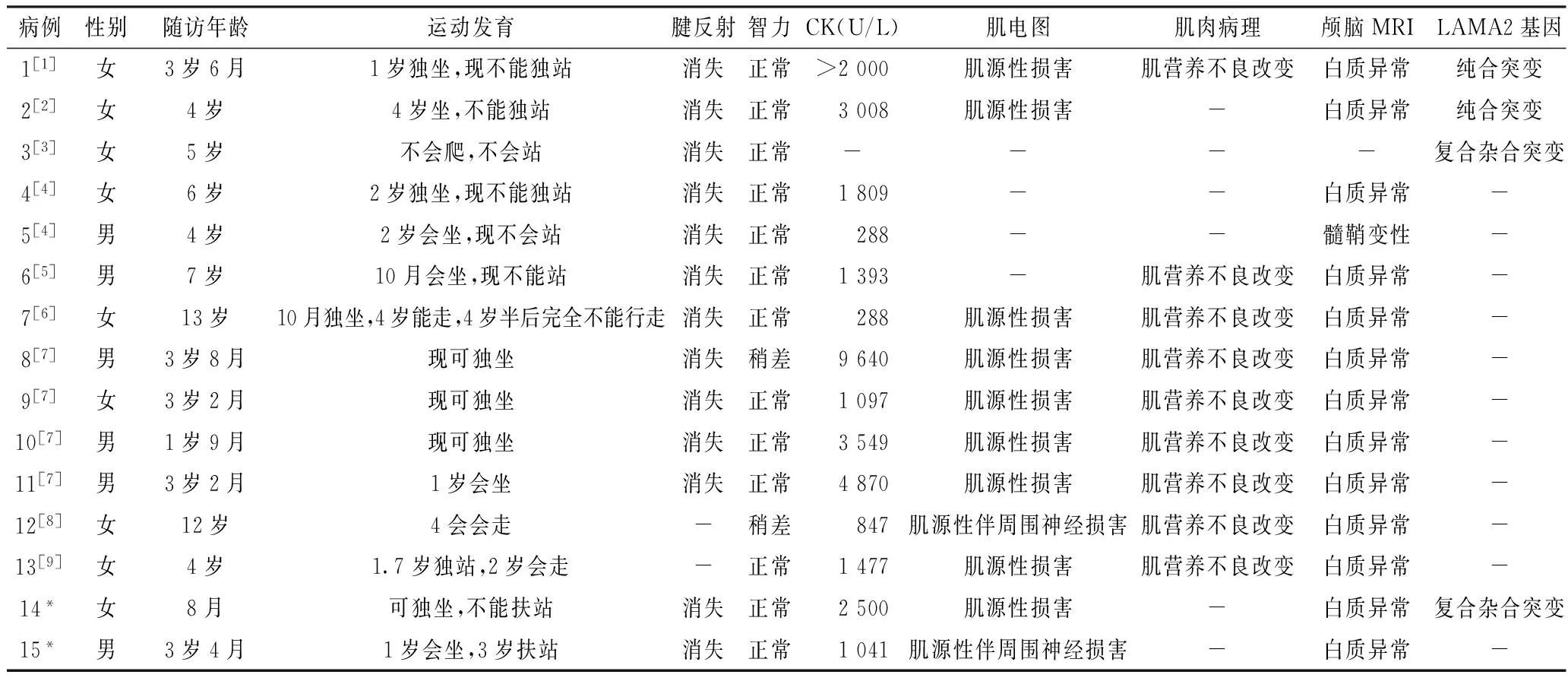
表1 15例MDC1A患儿的临床资料
注:*为本文病例,-为无此项;CK=肌酸激酶
因此,本文认为对因肢体无力就诊的患儿,若发现激酶谱明显升高,肌电图提示肌源性损伤改变,应常规进行颅脑MRI的检查,若存在脑白质营养不良的改变,应高度考虑MDC1A。
3.3 基因改变 MDC1A由LAMA2基因突变所致,基因定位在6q22-23,含65个外显子,突变覆盖9 500 bp的编码序列。突变类型包括无义、错义、缺失、重复以及剪接位点的改变。近些年欧美对其基因突变的类型做了大量的研究。2008年Oliveira等[12]对来自于葡萄牙、西班牙、瑞士等国家的26例患儿进行基因分析,发现18种基因突变;2010年Geranmayeh等[13]对51例患儿进行基因分析,发现44种基因的突变类型。由于遗传异质性的存在,同一种突变类型临床表现可以相差甚远[14],其不同突变类型与表型的关系尚需进一步研究。
本文先证者1进行LAMA2基因检测,因LAMA2基因庞大,突变类型多样,直接测序费用高,本研究采用二代测序法,结果在LAMA2基因上发现2个位点的杂合突变,一个为无义突变,致病性之前已有报道[13],编码区第4048号碱基由C变为T,导致本该编码精氨酸的1350号密码子变为终止密码子;第二个突变为移码突变,在编码区457号碱基和458号碱基之间插入了T,导致第153号密码子后发生移码变异,为国际上尚未报道的新发突变。本例发现的基因突变虽不是等位基因的两个相同位点上发生的纯合突变,但两个杂合突变均发生在LAMA2基因,且均为致病性突变,可以导致发生MDC1A的表型。本例因故未能对先证者父母进行基因检测,因先证者父母表型正常,MDC1A为常染色体隐性遗传,推测此两个突变分别来自父亲、母亲,即其父母分别各携带一个杂合突变而不致病,符合常染色体隐性遗传的规律。这种遗传方式既往曾有报道[3]。
15例患儿中有4例进行基因检测,均发现LAMA2基因突变,其中2例为纯合突变,2例为复合杂合突变,均符合常染色体隐性遗传的规律。说明基因检测对该病诊断具有确定意义。
该病目前无特效治疗,临床只能给予对症处理,改善功能缓解病情进展。有关动物实验的研究近年来较多,如应用层黏连蛋白111[15]、血管紧张素Ⅱ的1型受体拮抗剂[16]、整合素α7[17]等改善病变肌肉的病理结构和提高生存能力,希望不久的将来能用于临床,改善患儿的生活质量。
MDC1A系罕见病,致死率、致残率高,肌肉活检可以用于先证者的诊断,但无法进行产前检查,因此尽量明确先证者的基因类型,指导优生优育、进行产前诊断是预防该病患儿出生的惟一手段。
1 王硕,熊辉,罗静,等.一个先天性肌营养不良1A型家系的临床、分子病理及遗传学研究[J].中华医学遗传学杂志,2010,27(1):13-16.
2 He Z,Luo X,Liang L,et al.Merosin-deficient congenital muscular dystrophy type 1A:A case report[J].Exp Ther Med,2013,6(5):1233-1236.
3 朱艳慧,喻长顺,王晓春,等.层黏连蛋白α2缺失型先天性肌营养不良患儿一例LAMA2基因突变分析[J].中华临床医师杂志:电子版,2013,7(13):5871-5874.
4 吴元重,徐方元,吴纲烈.先天性肌营养不良-非福山型2例[J].安徽医学,2000,21(1):63.
5 龙冬兰,蔡少华,林茂增,等.Merosion 缺失型先天性肌营养不良一例[J].中华医学遗传学杂志,2005,22(4):469.
6 刘建国,沈定国.Merosin蛋白缺陷性先天性肌营养不良一例[J].中华神经科杂志,2006,39(4):263.
7 熊晖,姚生,袁云,等.先天性肌营养不良的诊断及层黏连蛋白表达的意义[J].中华儿科杂志,2006,44(12):918-923.
8 朱雯华,赵重波,林洁,等.先天性肌营养不良1A型的临床表现和病理改变(附1例报道)[J].中国临床神经科学,2008,16(5):504-508.
9 闫炳苍,李建军.先天性肌营养不良合并脑白质营养不良附1例报道[J].罕少疾病杂志,2010,17(1):50-51.
10 Quijano-Roy S,Renault F,Romero N,et al.EMG and nerve conduction studies in children with congenital muscular dystrophy[J].Muscle Nerve,2004,29(2):292-299.
11 刘茜玮,肖江喜,谢晟,等.MRI对先天性肌营养不良的诊断价值[J].临床放射学杂志,2010,29(4):506-509.
12 Oliveira J,Santos R,Soares-Silva I,et al. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients[J].Clin Genet,2008,74(6):502-512.
13 Geranmayeh F,Clement E,Feng LH,et al.Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations[J].Neuromuscul Disord,2010,20(4):241-250.
14 Di Blasi C,Bellafiore E,Salih MA,et al.Variable disease severity in Saudi Arabian and Sudanese families with c.3924 + 2 T > C mutation of LAMA2[J].BMC Res Notes,2011,4:534.
15 Rooney JE,Knapp JR,Hodges BL,et al.Laminin-111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin-deficient congenital muscular dystrophy[J].Am J Pathol,2012,180(4):1593-1602.
16 Meinen S,Lin S,Ruegg MA.Angiotensin Ⅱ type 1 receptor antagonists alleviate muscle pathology in the mouse model for laminin-α2-deficient congenital muscular dystrophy(MDC1A)[J].Skelet Muscle,2012,2(1):18.
17 Doe JA,Wuebbles RD,Allred ET,et al.Transgenic overexpression of the α7 integrin reduces muscle pathology and improves viability in the dy(W) mouse model of merosin-deficient congenital muscular dystrophy type 1A[J].J Cell Sci,2011,124(Pt 13):2287-2297.
