基于RNA-Seq的羊种布鲁氏菌新转录本与非编码RNA鉴定
2015-06-24郭英飞王玉飞龚春丽杨明娟袁久云庄妤冰柯跃华杜昕颖汪舟佳陈泽良
郭英飞,王玉飞,龚春丽,杨明娟,袁久云,庄妤冰,柯跃华,杜昕颖,汪舟佳,陈泽良
基于RNA-Seq的羊种布鲁氏菌新转录本与非编码RNA鉴定
郭英飞1,3,王玉飞2,龚春丽1,杨明娟1,袁久云1,庄妤冰1,柯跃华1,杜昕颖1,汪舟佳1,陈泽良1
目的 对羊种布鲁氏菌的转录本进行测序,鉴定基因组中新的转录本和非编码RNA。方法 提取羊布鲁氏菌的总RNA,去除rRNA后连接接头逆转录成cDNA,PCR扩增后进行测序,以羊布鲁氏菌16M的基因组序列为参照对测序得到的reads进行比对作图,通过生物信息学方法进行新转录本和非编码RNA的鉴定。结果 测序数据分析显示,reads在基因组上覆盖度较好。与现有注释基因相比,有773个基因的5′或3′端在基因组的原有位置基础上发生了延伸,并发现了16个新的转录本。根据测序结果,共鉴定出241条候选的非编码RNA(sRNA),进一步的RT-PCR验证结果显示,预测的sRNA在体外条件下有表达。结论 布鲁氏菌基因组中存在除了预测以外的新的转录本,布鲁氏菌中存在非编码RNA且在不同条件下有差异表达。
布鲁氏菌;RNA-Seq;转录组;sRNA
布鲁氏菌病(Brucellosis, 简称布病)是由布鲁氏菌引起的一种人兽共患传染病,危害严重,在世界各地都有广泛流行,该病不仅危害人类健康,还影响畜牧业的发展。目前布鲁氏菌可分为9种,即羊种、牛种、猪种、犬种、绵羊附睾种、沙林鼠种、田鼠种、鲸型海洋种和鳍型海洋种[1-2]。在我国流行的主要是羊、牛和猪种布鲁氏菌,其中羊种布鲁氏菌流行最为广泛。布鲁氏菌是一种胞内寄生菌,胞内生存和复制是布鲁氏菌的主要毒力特征。布鲁氏菌主要是通过改变吞噬体的成熟过程来避免吞噬体与溶酶体的融合,从而使得细菌可以到达其胞内复制部位内质网,开始大量复制[3]。基因组测序与基因的功能注释,对于深入理解布鲁氏菌的基因功能及其在致病性中的作用具有重要意义。
转录组是特定细胞或组织在特定时间或功能状态下转录出来的所有RNA的集合,是连接基因组遗传信息与蛋白质组生物功能之间的纽带[4-5]。随着新一代高通量测序技术的发展,转录组测序(RNA sequencing,RNA-Seq)技术已成为转录组研究的重要手段。RNA-seq能够对特定细胞或组织在某一环境或生理条件下的几乎所有转录本进行检测,它在分析转录本的表达水平、研究结构变异的同时,还能够发现未知转录本和非编码RNA[6-8]。与传统的芯片杂交技术相比,RNA-Seq无需设计探针,也不需要事先知道参考基因组序列的注释信息,并且具有分辨率高、背景噪音低等优势,目前已广泛应用于原核和真核生物的转录组学研究。本研究以羊布鲁氏菌为例,应用RNA-Seq技术对其进行高通量转录组测序分析,描绘出羊布鲁氏菌的转录组图谱,鉴定新的转录本以及非编码RNA,为深入理解布鲁氏菌与宿主细胞相互作用的机制奠定基础,同时也为进一步完善布鲁氏菌基因结构信息及挖掘潜在的新基因和非编码RNA提供了有价值的数据。
1 材料与方法
1.1 菌株、培养基及主要试剂
1.1.1 菌株 羊布鲁氏菌Brucellamelitensis16M为本室保存。
1.1.2 培养基及培养条件 大豆胰蛋白胨琼脂(TSA)和大豆胰蛋白胨肉汤(TSB)培养基购自生物梅里埃公司。挑取TSA平板上的羊布鲁氏菌单菌落在5 mL TSB过夜培养至后,37 ℃培养至稳定期,然后以1∶50转接到100 mL的TSB培养基中,培养至对数生长中期。
1.1.3 分子生物学试剂 MasterPureTMRNA Purification Kit购自Epicenter公司,DNase购于美国 Qiagen公司;SYBR Green I定量PCR试剂盒购自TaKaRa公司;Trizol 购自Invitrogen公司;MICROBExpress bacterial mRNA enrichment kit购自美国Ambion 公司;RNA连接酶、CIP 酶、PNK酶购于NEB公司;接头和引物在美国IDT公司合成;切胶回收试剂盒购于美国Qiagen公司;测序试剂购于美国Illumina公司。
1.2 RNA抽提和rRNA去除 MasterPureTMRNA Purification Kit提取羊布鲁氏菌的总RNA,并用DNase消化,方法参照试剂盒说明进行。之后,用MICROBExpress bacterial mRNA enrichment kit从总RNA中去除rRNA,纯化的mRNA通过Agilent 2100生物分析仪进行质量检测。
1.3 cDNA测序文库的构建及RNA-seq高通量测序 mRNA打断成100-500 nt短片段后逆转录成cDNA,3′末端加polyA并连接测序接头,连接产物经PCR扩增得到测序文库,用Illumina HiSeq 2000测序仪对文库进行序列测定。
1.4 转录组数据分析 测序所得的原始数据称为原始序列(raw reads)。原始序列去除接头序列、空读序列(N的比例大于10%的读数)以及低质量序列(质量值Q≤20的碱基数占整个读数的40%以上)后得到测序序列(clean reads)用于后续分析。参考基因组16M(AE008917.1和AE008918.1)及它们的注释基因信息均从NCBI数据库下载。使用短reads比对软件SOAPaligner/SOAP2软件将过滤后的clean reads比对到布鲁氏菌基因组和相关基因,通过比对结果统计reads在参考基因组及基因序列上的分布情况及覆盖度,同时利用RPKM(Reads Per Kb per Million reads)法[10]计算基因的表达量。
基因结构优化及新转录本的预测如图1所示,首先利用cufflinks软件将比对上羊布鲁氏菌基因组的测序序列进行组装拼接。通过比较gene model与现有注释基因的差别,对基因的5′和3′端进行延伸,由此优化基因结构。如果组装的转录本序列未能与现有基因比对上,而是位于现有基因之间的基因组上,同时满足下列条件:距离现有的注释基因200 bp以上;长度不短于150 bp;平均覆盖度大于2, 则这些序列有可能为潜在的新转录本及新基因。
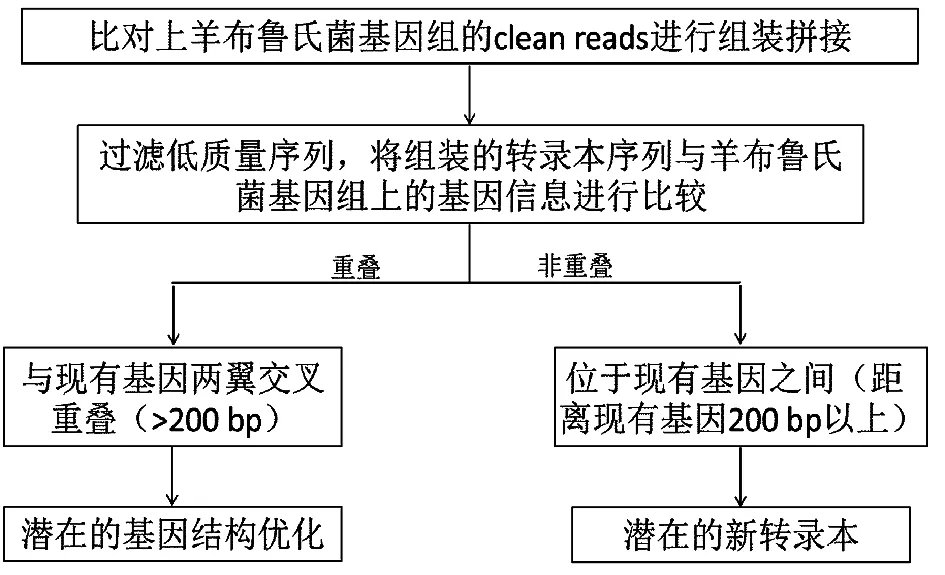
图1 基因结构优化及新转录本鉴定流程
非编码RNA(small non-coding RNA,sRNA)的预测主要是参考文献9,从潜在的gene model中挑选出长度大于等于100 bp且平均覆盖深度不小于的2的gene model,再从中找出位于基因间区的潜在gene model作为新转录本。如果其表达量与两翼编码区域相差15%以上,而且向上游或下游延伸200 nt有启动子的存在,则可作为候选的sRNA。
1.5 sRNA的RT-PCR验证 挑取候选的sRNA分子进行RT-PCR验证。将培养至对数生长中期的16M菌液离心,PBS漂洗1遍。将菌体重悬到等量的不同培养基中进行刺激处理后,用Trizol试剂提取布鲁菌的RNA。酸刺激是将菌体重悬到pH4.0的TSB培养基中,37 ℃作用20 min;高氧刺激是将菌体重悬到含有440 mmol/L H2O2的TSB培养基(pH7.0)中,37 ℃作用20 min;营养缺乏刺激是将菌体重悬到GEM培养基[10]中(pH7.0),37 ℃作用20 min;同时将菌体重悬于pH7.0的TSB培养基中,37 ℃作用20 min作为未处理对照。RNA反转录成cDNA后用针对候选sRNA分子和16sRNA的引物进行RT-PCR验证。
2 结 果
2.1 转录组的测序数据评估 利用琼脂糖凝胶电泳和Agilent 2100对提取的羊布鲁氏菌总RNA质量进行评估,检测结果显示5S、16S和23S RNA条带完整,23S/16S >1.5,OD260/280≥1.8,OD260/230≥1.8。结果表明RNA纯度较高,质量满足下一步建库要求。此外,我们通过检测reads在基因组上的分布来评价测序文库的随机性。统计参考基因上不同位置比对上的Reads数量,因为不同的参考基因有不同的长度,我们把 Reads在基因上的位置标准化到相对位置(Reads在基因上的位置与基因长度的比值)。结果表明reads在基因组上分布均匀(图2A),说明片段的随机性好。基因覆盖度统计表明,覆盖率达90%~100%的基因数有2 779条,约占88%;覆盖率达80%~90%的基因数有253条,约占8%(图2B)。此结果表明,我们的测序结果随机性好,并且对基因的覆盖度良好,数据可以用于后续的转录组分析。
2.2 基因结构的优化 基因编码区的起始与终止位置,是通过基因组注释得到的,但真正的起始与终止位置有待验证。通过转录组测序,能够对基因的结构进行进一步的确认与优化。基因结构优化分析显示,羊布鲁氏菌基因组上共有773个基因的5′和或3′端在原有基础上发生了延伸,不同延伸类型的统计结果见表2。从表中可以看出,I号染色体和II号染色体上的延伸分布有差异。部分基因的5′端和3′端的延伸情况见表3。
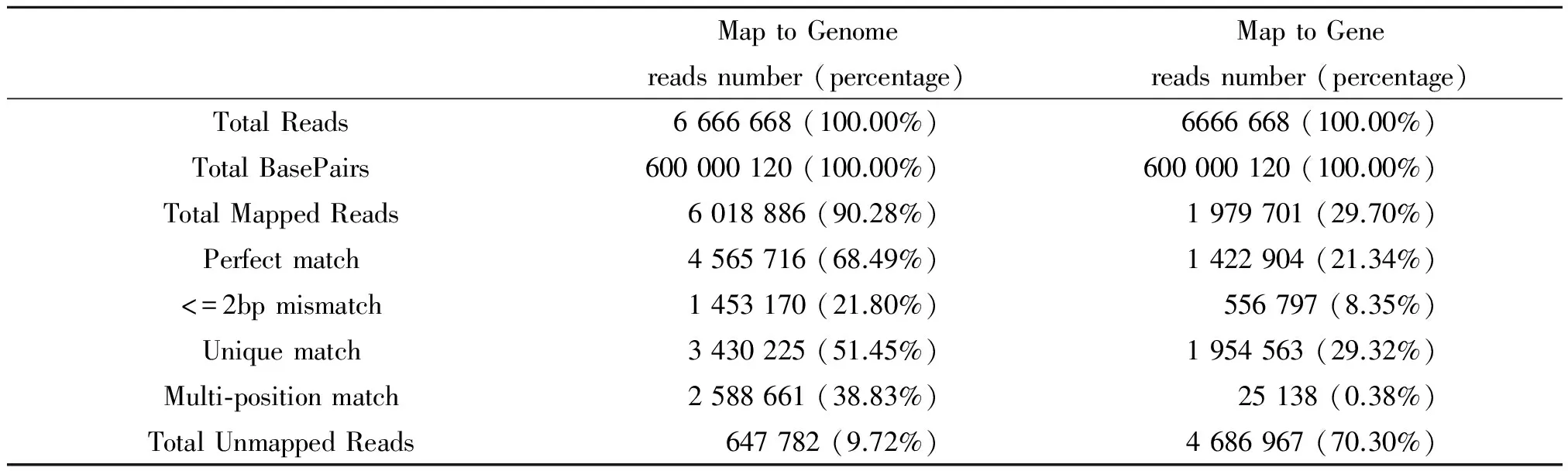
表1 样品和参考基因组及参考基因比对的统计结果
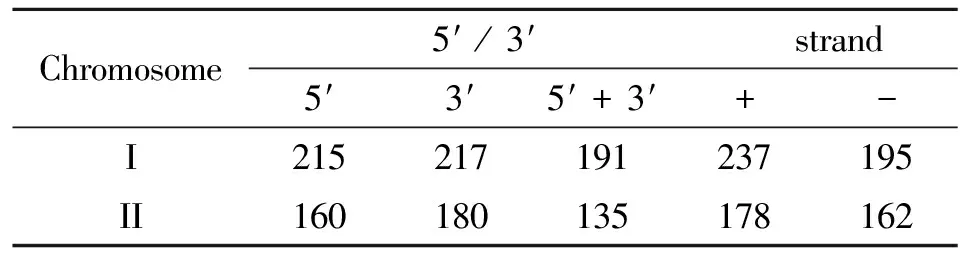
表2 基因起始与终止位置延伸的基因分布
2.3 新转录本的鉴定 通过RNA-seq,本研究共鉴定出16个新转录本,长度分布为166~748 bp,其中13个位于I号染色体上,3个位于II号染色体上,具体信息见表4。
2.4 sRNA的预测及鉴定 通过转录组测序,在羊布鲁氏菌16M的两条染色体上共筛选出241条候选的sRNA分子,大小在100~834 nt之间(表5),其中168个位于I号染色体上,73个位于II号染色体上。我们从中随机抽取了10个sRNA候选分子用RT-PCR进行了验证,结果表明其中7个sRNA分子在羊布鲁氏菌中存在转录本,而且大多数sRNA在不同的环境压力条件下表达量不同(图3),提示这些sRNA有可能在布鲁氏菌适应环境压力中发挥一定的作用。

图2 Reads随机性分析(A)及基因覆盖度统计(B)

表3 部分基因5′端和3′端的延伸
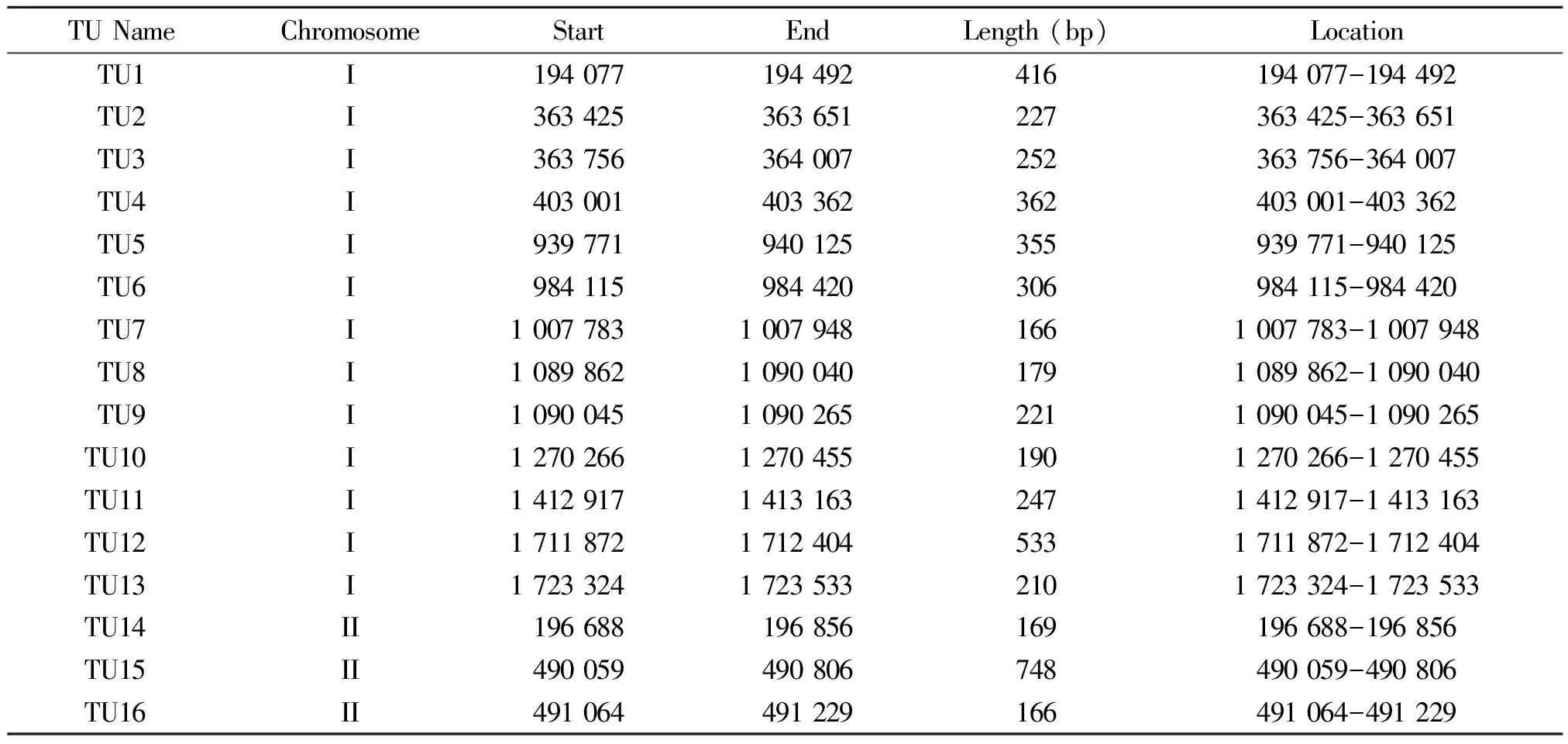
表4 鉴定的新转录本信息
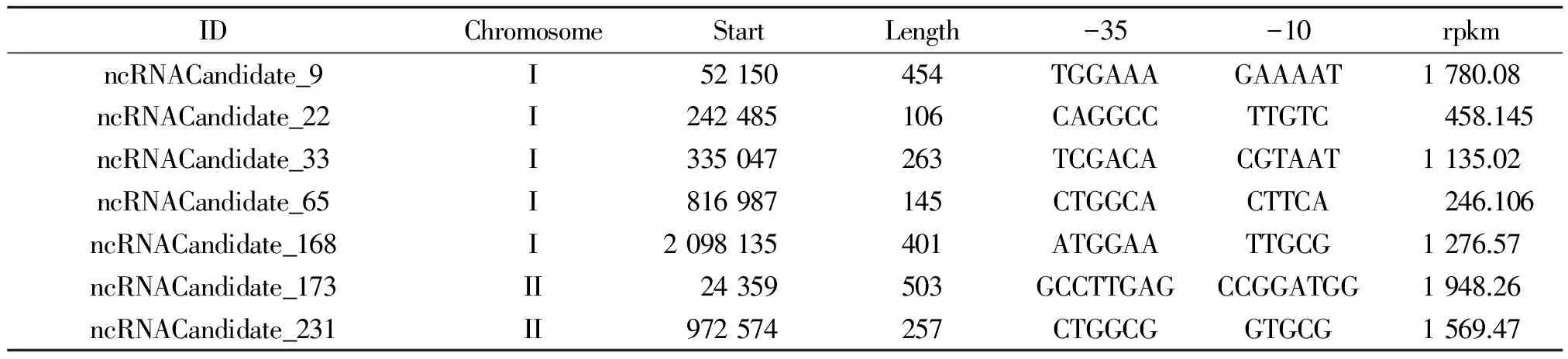
表5 部分鉴定的新sRNA信息
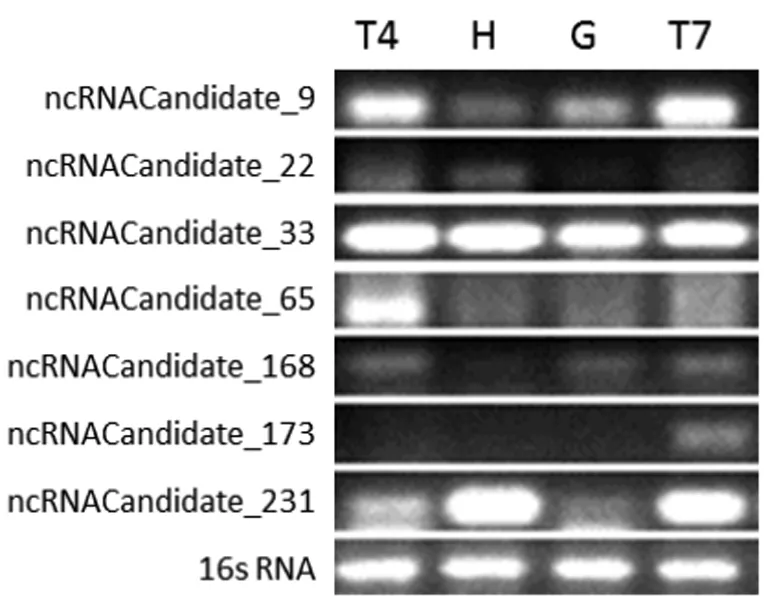
T4: TSB 4.0, H: H2O2,G:GEM 7.0,T7: TSB 7.0.
3 讨 论
布鲁氏菌是一种胞内寄生菌,适应胞内环境是布鲁氏菌致病与生存的关键,因此了解布鲁氏菌在胞内极端苛刻环境下的生存机制对理解其致病机制至关重要。目前,已完成布鲁氏菌7个种18株菌的全基因组测序工作,为进一步在分子水平研究布鲁氏菌与宿主相互作用奠定了基础。基于Solexa、454和SOLID平台的RNA-Seq作为近年来新发展起来的高通量转录组测序技术,为转录组学研究提供了一种方便、有效的手段。目前该技术已广泛用于基因表达水平比较分析,同时,该技术在进一步完善基因组结构信息、鉴定新转录本及非编码RNA方面的应用也受到大家的关注[11-14]。本研究应用RNA-Seq技术对羊布鲁氏菌进行高通量转录组测序分析,为更全面地理解其发病机制提供必需的基础资料。
对于RNA-Seq来说,没有基因组DNA污染的高质量RNA是保证其后续测序结果质量的关键环节。由于mRNA仅占到总RNA的3%~5%,因此当总RNA提取完成后需要进行mRNA富集。将富集后的mRNA打断成小片段,并对末端进行接头修饰,为防止RNA自连,我们对RNA的5′ 末端进行去磷酸化,然后再连接接头逆转录成cDNA,PCR扩增后进行测序。在构建cDNA文库时,采用特定的处理使其在后续的信息分析中能够区分转录方向,并且确定转录本的边界,从而可以更为准确的计算基因的表达量,同时挖掘sRNA及其它非翻译区的信息,获得基因表达谱芯片技术无法获得的信息。经Illumina高通量深度测序后, 我们得到了一个包含6 666 668条原始测序读数的测序文库。比对分析显示,能比对上参考基因组的reads所占比例为90.28%,其中比对到唯一位置的reads所占比例为51.45%。单位置比对(unique mapped)的reads主要为mRNA,而多位置比对(multi-position mapped)的reads基本上是rRNA和tRNA,因此我们在后续分析中使用的都是unique mapped reads,以保证分析结果的可信度。羊布鲁氏菌16M两条染色体上共有3 199条基因,3 170条基因被覆盖比对上,其中高峰度表达的基因(比对到基因中的reads数多于10的基因)有3 157条。此结果表明,正常羊布鲁氏菌至少表达转录了3 170条基因,而且绝大多数是高峰表达的。
RNA-Seq在进一步完善基因组结构信息和鉴定新转录本方面也发挥着重要作用。将RNA-Seq测序结果与羊布鲁氏菌16M现有基因组上基因进行比对后,发现共有773个基因的5′或3′端在原有基础上发生了延伸。该结果表明,原基因的5′或3′UTR区的预测存在一定的偏差,而这些延伸进一步优化了相关基因的结构。我们还对没有与现有基因比对上,而是位于现有基因之间的基因组上的转录本序列进行了分析,共发现了16个新的转录本,长度为166~748 bp,后续的深入研究需要对这些新转录本进行进一步的实验验证。
转录组测序的另一个重要作用就是挖掘和发现新的非编码小RNA。细菌基因组中除了编码基因外,还有一些不编码基因也可被转录出来。由于这类RNA长度较短,具有调控功能,也被称为非编码小RNA(small non-coding RNA,sRNA)。细菌sRNA的长度在50~500碱基之间,不编码蛋白质,常位于基因间区,也有少数位于反义链上[15]。目前已在多种细菌中发现了sRNA的存在,其中研究最广泛的是大肠杆菌和沙门氏菌。sRNA在细菌的生物学功能中发挥了重要的调节作用。目前认为大部分的细菌sRNA在应对环境变化的基因表达调控中发挥重要作用,是细菌适应环境压力的重要调控子。它们的主要功能是感应环境的变化,调控细胞的代谢途径、生长方式使之与环境相适应,以及控制细菌毒力基因的表达[16]。我们通过RNA-Seq在布鲁氏菌16M的两条染色体上共发现了241条候选的sRNA分子,大小在100~834 nt之间。这说明sRNA在布鲁氏菌中是普遍存在的。布鲁氏菌是一种胞内寄生菌,它们主要寄生于机体的单核细胞(主要是巨噬细胞)内。巨噬细胞内微环境包含有多种杀(抑)菌物质,比如酸、过氧化物、溶菌酶等。布鲁氏菌必须适应一系列不同的环境状态才能在宿主体内长期生存,而这种适应主要是通过调控基因的协调表达来实现的[17]。我们随机挑取了10个sRNA候选分子用RT-PCR进行了验证,结果表明其中7个sRNA分子在羊布鲁氏菌中均存在转录本,表明通过RNA-Seq的方法来发现sRNA是非常有效的。此外,我们还分析了这7个sRNA在不同的环境压力条件下的表达,RT-PCR结果表明除了ncRNACandidate_33外,其它sRNA在不同的刺激条件下表达量不同,说明这些sRNA有可能在布鲁氏菌适应胞内环境中发挥一定的作用。
综上所述,我们将RNA-Seq技术成功地用于布鲁氏菌的转录组研究中,该结果为描绘布鲁氏菌的转录组图谱奠定了基础。通过本研究,还优化了布鲁氏菌的基因结构、发现了新的转录本和新的sRNA,这些结果不仅增加了对布鲁氏菌现有基因组信息的注释,也为更全面地理解其致病机制奠定了基础。
[1]Chain PS, Comerci DJ, Tolmasky ME, et al. Whole-genome analyses of speciation events in pathogenic Brucellae[J]. Infect Immun, 2005, 73(12): 8353-8361. DOI: 10.1128/ IAI.73.12.8353-8361.2005
[2]Corbel MJ. Brucellosis: an overview[J]. Emerg Infect Dis, 1997, 3(2): 213-221. DOI: 10.3201/eid0302.970219
[3]Gorvel JP, Moreno E.Brucellaintracellular life: from invasion to intracellular replication[J]. Vet Microbiol, 2002, 90(1-4): 281-297. DOI: 10.1016/S0378-1135(02) 00214-6
[4]Hou ZW, Wang Z, Gao H, et al. The principle of dRNA-seq and its applications in prokaryotic transcriptome analyses[J]. Hereditas, 2013, 35(8): 983-991. (in Chinese) 侯志伟, 王赞, 高宏,等. dRNA-seq原理及其在原核生物转录组学研究中的应用[J]. 遗传, 2013, 35(8): 983-991.
[5]Gustincich S, Sandelin A, Plessy C, et al. The complexity of the mammalian transcriptome[J]. J Physiol, 2006, 575(Pt 2): 321-332. DOI: 10.1113/ jphysiol.2006.115568
[6]Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics[J]. Nat Rev Genet, 2009, 10(1): 57-63. DOI: 10.1038/nrg2484
[7]Tariq MA, Kim HJ, Jejelowo O, et al. Whole-transcriptome RNAseq analysis from minute amount of total RNA[J]. Nucleic Acids Res, 2011, 39(18): e120. DOI: 10.1093/nar/gkr547
[8]Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome[J]. Nat Biotechnol, 2011, 29(7): 644-652. DOI: 10.1038/nbt.1883
[9]Yoder-Himes DR, Chain PS, Zhu Y, et al. Mapping theBurkholderiacenocepacianiche response via high-throughput sequencing[J]. Proc Natl Acad Sci U S A, 2009, 106(10): 3976-3981. DOI: 10.1073/pnas.0813403106
[10]Wang YF, Qiao F, Zhong ZJ, et al. Transcriptional analysis ofBrucellavirulence regulation genes under stress conditions and during cell infection[J]. Chin J Microbiol Immunol, 2008, 28(10): 919-924. (in Chinese) 王玉飞, 乔凤, 钟志军. 布鲁菌重要毒力调控基因在环境刺激和细胞侵染过程中的转录研究[J]. 中华微生物学和免疫学杂志, 2008, 28(10): 919-924.
[11]Leyn SA, Kazanov MD, Sernova NV, et al. Genomic reconstruction of the transcriptional regulatory network inBacillussubtilis[J]. J Bacteriol, 2013, 195(11): 2463-2473. DOI: 10.1128/JB.00140-13
[12]Kroger C, Dillon SC, Cameron AD, et al. The transcriptional landscape and small RNAs ofSalmonellaentericaserovarTyphimurium[J]. Proc Natl Acad Sci U S A, 2012, 109(20): E1277-1286. DOI: 10.1073/pnas.1201061109
[13]Mutz KO, Heilkenbrinker A, Lonne M, et al. Transcriptome analysis using next-generation sequencing[J]. Curr Opin Biotechnol, 2013, 24(1): 22-30. DOI: 10.1016/j.copbio.2012.09.004
[14]Reddy JS, Kumar R, Watt JM, et al. Transcriptome profile of a bovine respiratory disease pathogen: Mannheimia haemolytica PHL213[J]. BMC Bioinformatics, 2012, Suppl 15: S4. DOI: 10.1186/1471-2105-13-S15-S4
[15]Gottesman S. Micros for microbes: non-coding regulatory RNAs in bacteria[J]. Trends Genetics, 2005, 21: 399-404. DOI: 10.1016/j.tig.2005.05.008
[16]Toledo-Arana A, Repoila F, Cossart P. Small noncoding RNAs controlling pathogenesis[J]. Curr Opinion Microbiol, 2007, 10(2): 182-188. DOI: 10.1016/ j.mib.2007.03.004
[17]Celli J. Surviving inside a macrophage: the many ways ofBrucella[J]. Res Microbiol, 2006, 157(2): 93-98. DOI: 10.1016/j.resmic.2005.10.002
Identification of novel transcripts and sRNA ofBrucellamelitensisby RNA-Seq
GUO Ying-fei1,3,WANG Yu-fei2,GONG Chun-li1,YANG Ming-juan1,YUAN Jiu-yun1,ZHUANG Yu-bing1, KE Yue-hua1,DU Xin-ying1,WANG Zhou-jia1,CHEN Ze-liang1
(InstituteofDiseaseControlandPrevention,AcademyofMilitaryMedicalSciences,Beijing100071,China)
To identify novel transcripts and sRNA in genome ofB.melitensisby transcriptome sequencing, total RNA were extracted fromB.melitensisculture and rRNA were removed. After the addition of adaptor, RNA was reversely transcribed into cDNA, which were then subjected to PCR amplification and sequencing. The generated reads were mapped to genome sequence ofB.melitensisstrain 16M. With the mapping results, novel transcripts and sRNA were identified by bioinformatics methods. Sequencing results analysis showed that genome sequence was covered with the reads with good quality. A total of 773 genes were extended in their 5′ and/or 3′ ends of their original locations. Sixteen novel transcripts and 241 sRNAs candidates were identified. RT-PCR showed that some of the sRNAs were differentially expressed under stress conditions. InB.melitensisgenome, there is novel transcript which is not predicted. The sRNA does exist inB.melitensisand were expressed under different conditions.
Brucellamelitensis; RNA-Seq; transcriptome; sRNA
s: Wang Yu-fei, Email: yufeiwang21@yahoo.com; Chen Ze-liang, Email: zeliangchen@yahoo.com
10.3969/cjz.j.issn.1002-2694.2015.03.006
国家自然科学基金(81171530),北京市科技新星资助项目(Z131102000413062),北京市自然基金项目(6122030)
王玉飞,Email:yufeiwang21@yahoo.com 陈泽良,Email:zeliangchen@yahoo.com
1.军事医学科学院疾病预防控制所,北京 100071; 2.武警总医院检验科,北京 100069; 3.空军司令部门诊部,北京 100843
R378.5
A
1002-2694(2015)03-0216-06
Supported by the National Natural Science Foundation of China (No. 81171530), the Beijing Novo Program (No. Z131102000413062), and the Natural Science Foundation of Beijing (No. 6122030)
2014-08-11;
2014-12-05
