两步水热法制备纳米花状Ni3Fe/Ni3S2 高效电催化剂促进碱性电解水析氧反应
2023-11-22陈宇翔何捍卫
陈宇翔,何捍卫
(中南大学 粉末冶金研究院,长沙 410083)
随着传统化石能源的消耗与新能源的推广,氢能因高能量密度、零污染、零排放等特点被开发应用[1]。众多制氢方法中,电解水制氢因环保无污染的特点在工业生产中被广泛应用。在整个电解水过程中,析氢反应(hydrogen evolution reaction, HER)发生在阴极,析氧反应(oxygen evolution reaction,OER)发生在阳极。OER 因涉及动力学缓慢的四电子转移过程,有较高的过电位,使得电解水的槽电压过高[2],使用OER 催化剂降低电解水的槽电压可减少电解水制氢的能耗[3]。贵金属基催化剂氧化钌(RuO2)与氧化铱(IrO2)都具有优异的OER 活性,但其价格昂贵导致无法广泛应用[4]。因此,开发低成本、工艺简单且具有优异OER 活性的非贵金属基催化剂是碱性电解水领域的研究热点。研究表明,成本低廉的Ni 基催化剂,如NiCo、NiFe、NiCu合金以及Ni 基氧化物、氢氧化物、磷化物和硫化物等,在碱性条件下具备一定的催化性能[5-6]。其中Fe 与Ni 同属过渡金属,具有相同的3d 电子结构,两者易产生协同效应提升催化活性,且Fe 相较于Cu、Co 虽然本征活性略微逊色,但价格低廉。HU 等[7]采用水热法在泡沫镍(nickel foam, NF)上制备FeNiOxHy纳米阵列,该样品在1 mol/L KOH 溶液中,1 000 mA/cm2的电流密度下的析氧过电位仅为306 mV,电解80 h 后过电位几乎没有衰减。有研究表明,通过阴离子调控NiFe 催化剂,可以进一步优化其本征活性。LI 等[8]通过溶剂热法合成NiFeS 纳米材料,通过改变S 源的用量调控催化剂活性位点的电子结构,提升NiFe 催化剂的本征导电性能。在NiFe 中引入S 元素还可能产生NiFe 基硫化物,其金属特性可加快NiFe 催化剂中电子转移的速度[9]。CHEN 等[10]采用水热法制备一种垂直排列的Fe-Ni-S/NF 电极,生成的Ni3S2吸附OH-的同时,促进NiFeOOH 在OER 过程中生成更多析氧活性相。ZHONG 等[11]通过三步法制备出(Ni,Fe)S2/NiFe-CNFs(carbon nanofibers, CNFs),由于NiFe-CNFs 与 (Ni,Fe)S2形成异质结构产生协同效应,测得该催化剂的OER 过电位仅为287 mV。
鉴于S 可以改善NiFe 催化剂的本征活性,本文向FeNiOxHy催化剂中引入S 的同时构筑异质结构,旨在提升FeNiOxHy前驱体的OER 活性。当前,合成NiFeS 系催化剂的方法主要有电沉积法[12]、水热法[13]、高温固相法[14]等。水热法相较于其他方法,制备出的NiFeS 系催化剂形貌独特、丰富立体,可以提高催化剂的空间利用率。与泡沫镍相比,镍网力学性能好且耐腐蚀[15]。本文采用两步水热法在镍网上制备纳米花状的Ni3Fe/Ni3S2催化剂,对其微观结构与电催化析氧性能进行研究,为制备具有高催化活性的NiFeS 系OER 电催化剂提供了一条新策略。
1 实验
1.1 试剂
本实验使用的试剂均购自上海国药化工有限公司,包括九水合硝酸铁(Fe(NO3)3·9H2O)、氟化铵(NH4F)、尿素(CH4N2O)、氢氧化钾(KOH)、九水合硫化钠(Na2S·9H2O)、氢氧化钠(NaOH)、硅酸钠(NaSiO3)、碳酸钠(Na2CO3)以及十二水合磷酸钠(Na3PO4·12H2O),以上所有化学试剂都为分析级。所用镍网(nickel mesh, NM)经过喷砂前处理,目数为40 目,该镍网同时起到镍源与基底的作用。选择雷尼镍电极作为对比,研究所制备样品的电催化活性。镍网与雷尼镍均由中国船舶集团有限公司第七一八研究所(邯郸)提供。实验用水是经Millipore系统净化的去离子水,电阻率为18.25 MΩ·cm。
1.2 制备方法
剪取尺寸为2 cm×5 cm 的矩形镍网,放入混合碱液(NaOH 5 g/L,NaSiO320 g/L,Na2CO310 g/L,Na3PO440 g/L)中除油,于60 ℃下浸泡30 min,随后用去离子水清洗干净,置于乙醇中备用。
将1.00 g 的Fe(NO3)3·9H2O、0.24 g 的NH4F 和0.75 g 的CH4N2O 加入到50 mL 去离子水中,在室温下溶解,得到混合溶液。将溶液磁力搅拌10 min后倒入100 mL 的水热反应釜,再将预处理后的镍网浸入溶液中。将反应釜放入鼓风干燥箱中,溶液发生水热反应,反应温度为120 ℃,时间为6 h,得到FeNiOxHy/NM 样品,用去离子水润洗干净,在60 ℃的真空干燥箱中干燥12 h。
取2.4 g 的Na2S·9H2O 加入到50 mL 去离子水中, 在室温下溶解,磁力搅拌20 min 后倒入100 mL的水热反应釜中,再将FeNiOxHy/NM 前驱体浸入溶液中。将反应釜放入鼓风干燥箱中,在100 ℃反应8 h,得到黑色的Ni3Fe/Ni3S2/NM 样品,用去离子水润洗样品表面,干燥完毕进行后续表征与电化学性能测试。图1 所示为Ni3Fe/Ni3S2/NM 的制备过程示意图。
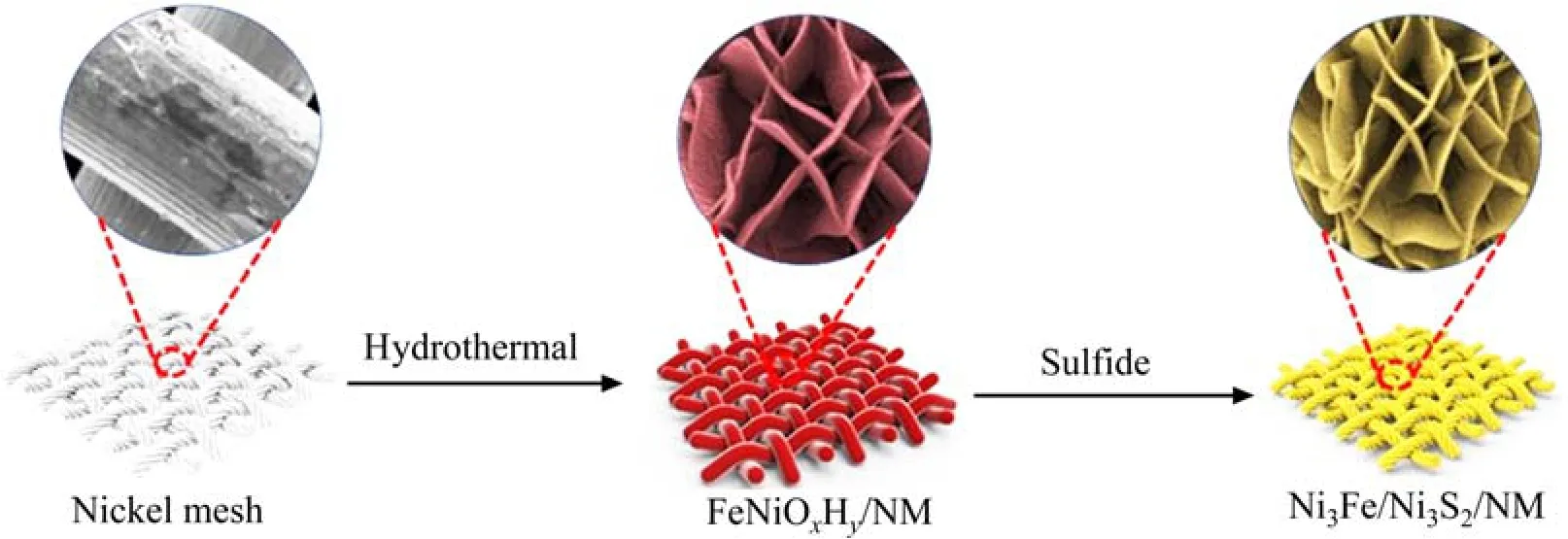
图1 Ni3Fe/Ni3S2/NM 的制备过程示意图Fig.1 Schematic diagram of the preparation process of Ni3Fe/Ni3S2/NM
1.3 样品表征
采用扫描电子显微镜(SEM, MIRA4 LMH,捷克)和能量色散X 射线谱仪(EDS, AXIS SUPRA)研究Ni3Fe/Ni3S2/NM 催化剂的微观形态和元素组成及分布。通过X 射线衍射仪(XRD, PANalytical Empyren)研究样品的晶体结构,靶材为 Cu 靶(λ=0.154 059 nm),管电压和电流分别为45 kV 和40 mA,扫描速度为10 (°)/min。催化剂表面的元素种类和化学价态通过 X 射线光电子能谱仪(XPS,Thermo Fisher Scientific, ESCALAB 250Xi, USA)测定,所用靶材为Al Kα(1 486.6 eV),并利用Avantage软件拟合每种元素的价态。
1.4 电化学测试
通过CHI660B 电化学工作站在100 mL 玻璃电解池中使用标准三电极系统进行电化学测试。分别在室温条件(25 ℃,1 mol/L KOH)与模拟工业条件(80 ℃,5.35 mol/L KOH)下测试Ni3Fe/Ni3S2/NM 催化剂的析氧性能,未经特殊说明默认测试条件为室温条件。将O2引入电解液使得溶液氧饱和,在测试前将整个标准三电极系统浸入电解液中200 s。将Ni3Fe/Ni3S2/NM 电极、石墨电极和饱和甘汞电极(saturated calomel electrode, SCE)分别作为工作电极、对电极和参比电极。极化曲线测试窗口E(vs SCE)为0~1.2 V,扫描速度为2 mV/s。上述电化学测试的电位,均通过Nernst 方程矫正为可逆氢电极(reversible hydrogen electrode, RHE),并且进行电位补偿:
式中:E(vs RHE)为工作电极相对于可逆氢电极的电位,E(vs SCE)为工作电极相对于饱和甘汞电极的电位,T为温度,I为电流,R为溶液阻抗。
通过循环伏安法(cyclic voltammetry, CV)测试Ni3Fe/Ni3S2/NM 的双电层电容(Cdl),在非法拉第区的扫描速率为10~100 mV/s。在电位E(vs RHE)为1.4~1.6 V 的范围内,进行6 000 次CV 循环测试,以评估样品的循环稳定性。电化学交流阻抗谱(electrochemical impedance spectroscopy, EIS)测试的电压E(vs RHE)为1.509 V,频率为105~10-2Hz,交流振幅为 5 mV。测量样品的计时电位(chronopotentiometry, CP)曲线,以反映电催化剂在稳定电流密度(10 mA/cm2)和多步连续电流密度(10~100 mA/cm2)下的稳定性。
2 结果与讨论
2.1 形貌与结构
图2(a)所示为镍网与Ni3Fe/Ni3S2/NM 的XRD谱图,Ni3Fe/Ni3S2/NM 谱图中存在明显的Ni3S2峰,在21.75°、31.10°、37.79°、49.73°和55.16°处的衍射峰分别为Ni3S2(JCPDS No. 44-1418)的(101)、(110)、(003)、(113)以及(122)晶面。镍网和Ni3Fe/Ni3S2/NM 的XRD 谱图都具有相似的3 个强峰,不同的是镍网基底的3 个强峰在44.4°、51.8°、76.3°处分别为Ni(JCPDS No. 70-1849)的(111)、(200)、(220)晶面,而Ni3Fe/Ni3S2/NM 的3 个主要衍射峰在44.2°、51.5°、75.8°处为Ni3Fe(JCPDS No. 88-1715)合金的(111)、(200)、(220)晶面[16],将XRD 谱图的3 个主峰放大,如图2(b)、(c)、(d)所示,可以明显看出Ni3Fe/Ni3S2的3 个主要衍射峰相较于镍网基底的衍射峰产生了负位移,这是由于Fe 原子取代Ni原子,引起位点间的晶格畸变而导致的晶格扩展造成的[17],位移后的衍射峰与Ni3Fe 的PDF 卡片对应,这证实了Ni3Fe/Ni3S2的成功合成。

图2 镍网基底和Ni3Fe/Ni3S2/NM 的XRD 谱图Fig.2 XRD patterns of Ni mesh substrate and Ni3Fe/Ni3S2/NM
通过XPS 对Ni3Fe/Ni3S2复合材料表面的相态组成进行进一步分析。图3(a)所示为Ni3Fe/Ni3S2复合材料的XPS 全谱图,可以看到Ni、Fe、O、S 以及C 的峰,其中C 峰来源于电荷校准处理,O 峰为催化剂在空气中氧化后的产物。图3(b)所示为Ni 2p的谱图,可以看到位于854.4 eV 和872.5 eV 处的2个强峰,分别对应Ni2+2p3/2和Ni2+2p1/2轨道,表明了Ni3S2的存在[18],861.4 eV 和878.4 eV 处的峰分别为854.4 eV 和872.5 eV 处强峰的卫星峰。858.4 eV 位置的峰对应Ni3+2p3/2,Ni 在空气中被氧化形成高价态Ni,而其右边851.8 eV 处强度较小的峰是典型的金属Ni 峰或Ni—S 键的特征峰[19]。图3(c)所示为Fe 2p 的谱图,707.1 eV 和719.8 eV 处的2个峰属于Fe02p3/2和Fe02p1/2,证明了金属态Fe 的存在[20]。位于711.5 eV 和724.5 eV 处的2 个峰分别代表Fe2+2p3/2和Fe2+2p1/2,剩下一组结合能为714.7 eV 和727.8 eV 的2 个峰分别代表Fe3+2p3/2和Fe2+2p1/2[21],金属态Fe 的存在表明Ni3Fe 合金的形成。图3(d)所示为S 2p 的谱图,163.1 eV 处的强峰再次证实了Ni3S2的存在[22],168.2 eV 位置的峰属于—SOx—,说明S 元素在空气中轻微氧化[23]。

图3 Ni3Fe/Ni3S2 的XPS 谱图Fig.3 XPS spectra of Ni3Fe/Ni3S2 (a), Ni 2p (b), Fe 2p (c), and S 2p (d) regions
图4所示为FeNiOxHy/NM 和Ni3Fe/Ni3S2/NM的扫描电镜图。图4(a)~(c)为FeNiOxHy/NM 在不同放大倍数下的微观形貌,可以看到FeNiOxHy以纳米球状生长在镍网基底上,这些纳米球由细微的纳米片堆叠而成,纳米片的厚度约为100~300 nm。图4(d)~(f)为Ni3Fe/Ni3S2/NM 的微观形貌,可见Ni3Fe/Ni3S2以纳米花状均匀生长在镍网基底上,这些纳米花由细小的纳米片堆叠形成,与FeNiOxHy前驱体相比,硫化过程使Ni3Fe/Ni3S2纳米片厚度变薄,约为50 nm,这极大地增加了催化剂的比表面,同时暴露出更多活性位点,有助于提高催化活性。

图4 FeNiOxHy/NM 和Ni3Fe/Ni3S2/NM 的SEM 图Fig.4 SEM images of FeNiOxHy/NM (a)–(c) and Ni3Fe/Ni3S2/NM (d)–(f)
图5(a)所示为Ni3Fe/Ni3S2/NM 的透射电镜图,Ni3Fe/Ni3S2/NM 的纳米片结构能明显被识别,厚度约为50 nm,这与图4(f)的扫描电镜结果一致。图5(b)、(c)、(d)为Ni3Fe/Ni3S2/NM 的高分辨透射电镜照片,观察到Ni3Fe 与Ni3S2共存。图5(c)清晰地显示了交错的Ni3S2晶格条纹,晶格间距分别为0.287、0.237 和0.234 nm,分别对应了Ni3S2的(110)、(003)和(021)晶面,Ni3S2的两个(110)晶面的夹角约为60°,说明热力学稳定的{001}晶面暴露了出来;(003)和(021)晶面之间的夹角约为70.7°,暴露出高折射率的{210}晶面,这有助于与Ni3Fe 的(111)晶面产生协同效应从而促进催化活性[9]。图5(d)中晶格条纹相对单一,其晶面间距为0.204 nm,对应Ni3Fe的(111)面。图5(e)为Ni3Fe/Ni3S2/NM 纳米片的选区电子衍射图,为经典的多晶衍射环。经过校准与比对,确定白色的衍射环对应Ni3S2的(021)、(110)与(003)晶面,黄色的衍射环对应Ni3Fe 的(111)晶面,这与HRTEM 结果相对应。图5(f)为Ni3Fe/Ni3S2/NM在透射电镜下的面扫能谱图,结果表明Ni、Fe、S元素均匀分布,元素含量如表1 所列,进一步证明了Ni3Fe/Ni3S2复合物的成功合成,并且形成了异质结构。
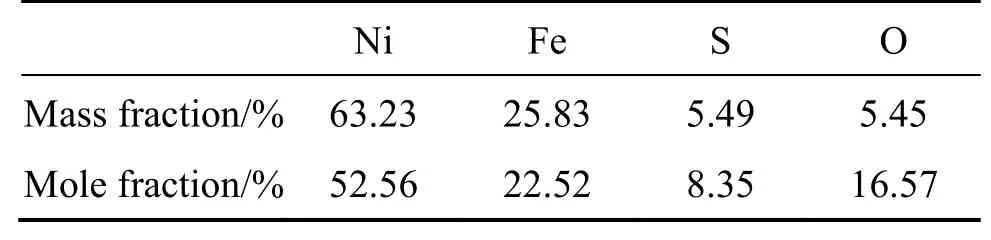
表1 Ni3Fe/Ni3S2/NM 的元素组成Table 1 The element composition of Ni3Fe/Ni3S2/NM

图5 Ni3Fe/Ni3S2/NM 的TEM 图、HRTEM 图、选区电子衍射图和能谱扫描图Fig.5 TEM image (a), HRTEM images (b)–(d), SAED pattern (e), and EDS element mapping (f) of Ni3Fe/Ni3S2/NM
2.2 电催化活性
分别在氧饱和的室温条件(25 ℃,1 mol/L KOH)以及模拟工业条件(80 ℃,5.35 mol/L KOH)下用标准三电极体系评估Ni3Fe/Ni3S2/NM 的析氧活性,并在相同的条件下测试FeNiOxHy/NM、雷尼镍与镍网的析氧活性作为对比,结果如图6 所示。图6(a)为4 种样品经过iR 校正后在室温条件下的阳极极化曲线,Ni3Fe/Ni3S2/NM 的极化曲线在1.4 V 处的氧化弱峰对应Ni2+被氧化成Ni3+的过程[24]。电流密度为10 mA/cm2时,Ni3Fe/Ni3S2/NM 的析氧过电位仅为229 mV,明显优于FeNiOxHy/NM(323 mV)、商业雷尼镍(305 mV)以及镍网(392 mV)。Ni3Fe/Ni3S2/NM电极的析氧活性普遍优于最近报道的NiFe 基析氧催化剂,如表2 所列。
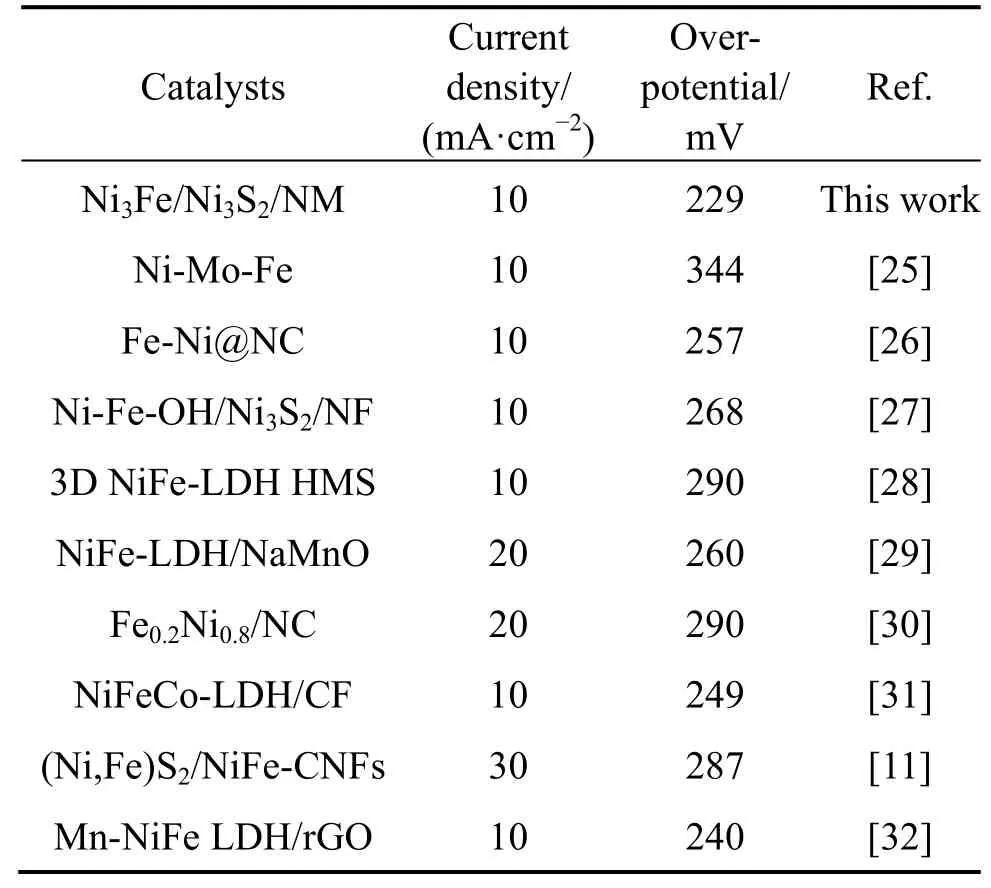
表2 NiFe 基催化剂的析氧活性Table 2 OER activity of NiFe-based catalysts
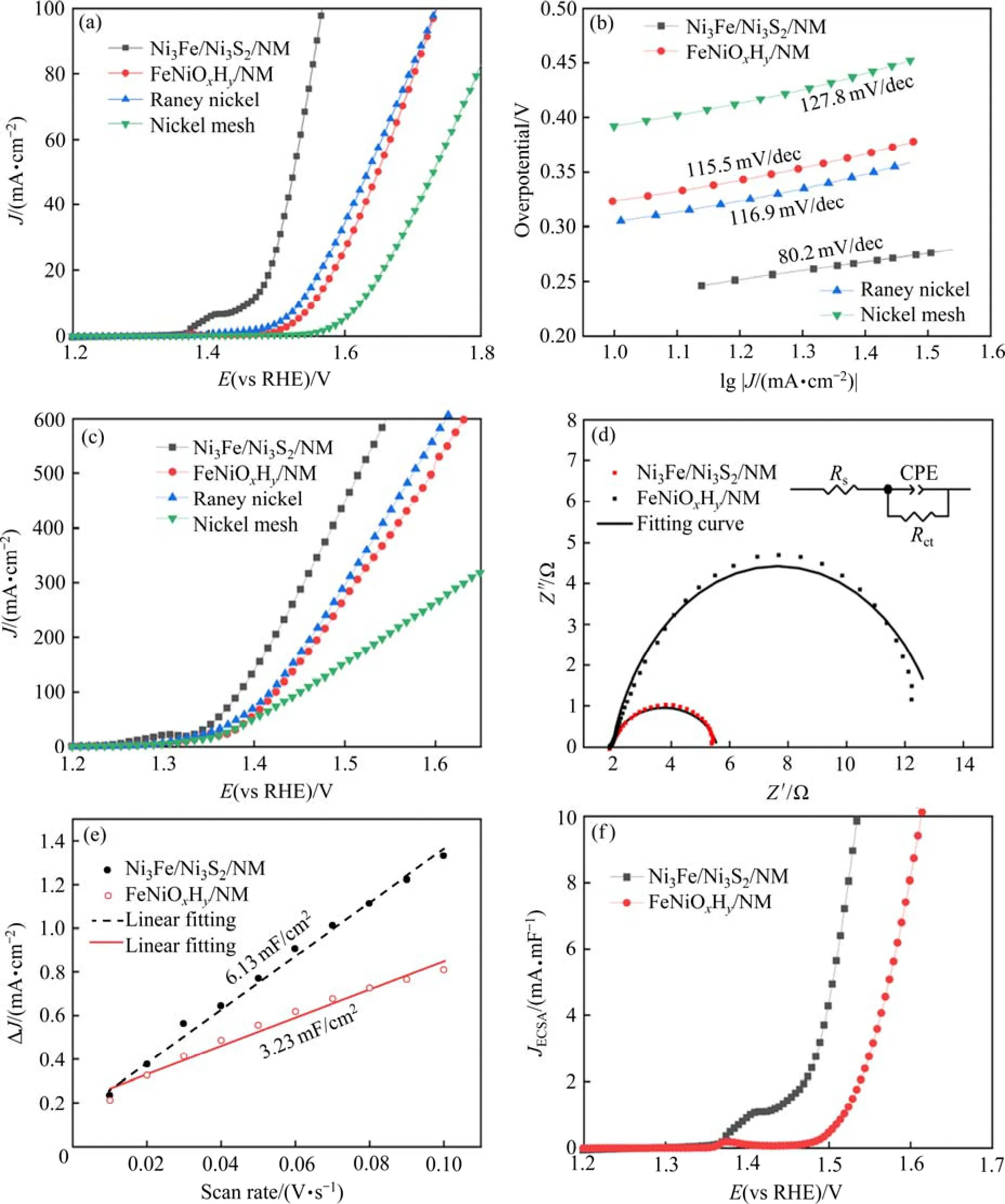
图6 Ni3Fe/Ni3S2/NM、FeNiOxHy/NM、雷尼镍和镍网的电化学性能Fig.6 Electrochemical properties of Ni3Fe/Ni3S2/NM, FeNiOxHy/NM, Raney nickel, and nickel mesh
Tafel 斜率是判断催化剂反应动力学的指标之一,由图6(b)可知,Ni3Fe/Ni3S2/NM 的Tafel 斜率最小,为80.2 mV/dec,低于FeNiOxHy/NM(115.5 mV/dec)、商业雷尼镍(116.9 mV/dec)与镍网基底(127.8 mV/dec)。Tafel 斜率越小,说明材料的析氧反应动力学越快,当增加相同的电流密度时,过电位增加值越小,说明Ni3Fe/Ni3S2/NM 越具有优异的OER 动力学过程。根据Krasil′shcikov 路径[33]:
式中:*代表活性位点。
从式(2)~(5)得出,碱性环境下的OER 机理包括OH-的电吸附过程、*—O 的形成与O2分子的形成。Ni3Fe/Ni3S2/NM 的Tafel 斜率为80.2 mV/dec,说明整个OER 过程的控制步骤是OH-的电吸附过程。Ni3Fe/Ni3S2/NM 的Tafel 斜率最低的主要原因可能是Ni3Fe 加速了OH-的吸附并转化成氧中间体,从而降低了氧中间体的吸附能[34]。
图6(c)所示为模拟工业条件下4 种样品的极化曲线。由图可知,Ni3Fe/Ni3S2/NM 的过电位在大电流密度下也很小,表现出优异的OER 活性,电流密度为600 mA/cm2时析氧过电位仅为335 mV,比商业雷尼镍(388 mV)的低53 mV,说明Ni3Fe/Ni3S2/NM 有工业应用的前景。
电化学阻抗谱(EIS)可以直观研究催化剂在OER 过程中的电荷转移动力学,图6(d)所示为Ni3Fe/Ni3S2/NM 与FeNiOxHy/NM 的电化学阻抗谱,插图为等效电路模型图,Rct为OER 过程中的电荷转移电阻,Rs为溶液电阻,CPE(constant phase element)为恒相位元件。在OER 过程中,Rct越小,表明电子转移越快,样品的OER 活性越好。拟合电路各元件数据如表3 所列,表中n为拟合出来的数值,越靠近1 代表拟合度越好。Ni3Fe/Ni3S2/NM与FeNiOxHy/NM 的Rct分别为3.70 Ω 与11.35 Ω,说明Ni3Fe/Ni3S2/NM 的电子转移速率更快,OER活性也更好。Ni3Fe/Ni3S2/NM 的电荷转移电阻小是因为在OER 过程中Ni3Fe 表面会生成OER 活性物质NiOOH 与FeOOH[35],这两者之间的协同效应会促进Ni3Fe/Ni3S2/NM 的OER 性能,同时内部的Ni3Fe 仍保持金属的导电性,Ni3S2具有良好的本征导电性,这些均有利于催化剂本身电子传输[36]。因此Ni3Fe/Ni3S2/NM 相较于FeNiOxHy/NM 具有更好的OER 活性。
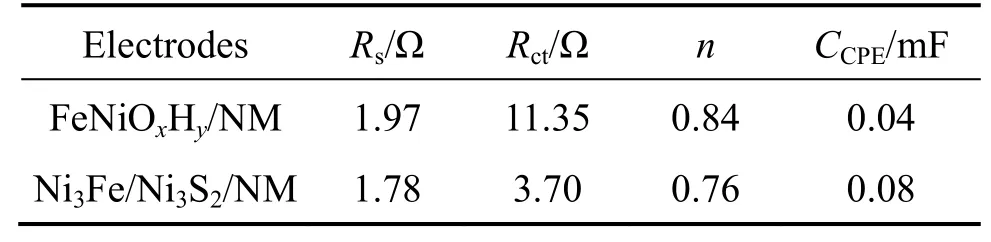
表3 Ni3Fe/Ni3S2/NM 和FeNiOxHy/NM 的EIS谱图拟合结果Table 3 Fitted paramaters of EIS spectra of Ni3Fe/Ni3S2/NM and FeNiOxHy/NM
电化学活性表面积(electrochemical active surface area, ECSA)表明电极表面活性位点的数量,先通过CV 曲线的非法拉第区域计算得到双电层电容(Cdl),再通过下式计算活性位点数目AECSA[37]:
式中:Cs代表该电极的比电容,镍铁基催化剂的取值一般为0.04 mF/cm2。
由于Cdl与AECSA成正比,因此可以用Cdl来比较催化剂的活性位点数目。如图6(e)所示,Ni3Fe/Ni3S2/NM 的Cdl(6.13mF/cm2)大于FeNiOxHy/NM 的Cdl(3.23 mF/cm2),所以Ni3Fe/Ni3S2/NM 电极表面析氧活性位点数目更多。图6(f)为经ECSA 归一化后Ni3Fe/Ni3S2/NM 与FeNiOxHy/NM 的阳极极化曲线,表征的是每一个活性位点平均的催化活性即本征催化活性,在相同的电流密度下Ni3Fe/Ni3S2/NM 的过电位小于FeNiOxHy/NM,即Ni3Fe/Ni3S2/NM 的本征催化活性更好。
除了催化活性外,稳定性也是评估催化剂性能的重要指标,对工业生产有重要的指导意义。图7(a)为Ni3Fe/Ni3S2/NM 在室温下的长时电解曲线,发现Ni3Fe/Ni3S2/NM 在10 mA/cm2的恒电流下电解60 h还可以保持稳定的析氧活性。图7(b)为Ni3Fe/Ni3S2/NM 在室温下的分步电解曲线,电解的电流密度从10 mA/cm2开始增加,每过500 s 升高10 mA/cm2,直至100 mA/cm2。从图看出,电压随电解电流密度的增加呈平稳的阶梯状增长,说明Ni3Fe/Ni3S2/NM催化电极具备良好的动态稳定性[38]。图7(c)为工业条件下,600 mA/cm2时Ni3Fe/Ni3S2/NM 经不同循环圈数后的极化曲线,从图中看出Ni3Fe/Ni3S2/NM在循环6 000 圈后的极化曲线与初始未循环的极化曲线几乎完全重合,通过计算得到Ni3Fe/Ni3S2/NM的过电位衰减率仅为2.39%。图7(d)所示为电流密度为600 mA/cm2时不同循环圈数下的过电位,可以看出Ni3Fe/Ni3S2/NM 在循环期间过电位较稳定。图7(e)所示为Ni3Fe/Ni3S2/NM 在工业条件下的长时电解曲线,随电解时间延长,Ni3Fe/Ni3S2/NM 析氧电位略有上升,在600 mA/cm2下电解24 h 后,Ni3Fe/Ni3S2/NM 过电位衰减率为2.9%,仍能保持97.1%的析氧活性。上述结果表明,在工业条件大电流密度下,Ni3Fe/Ni3S2/NM 电极的OER 性能优异,具备一定的工业应用潜力。
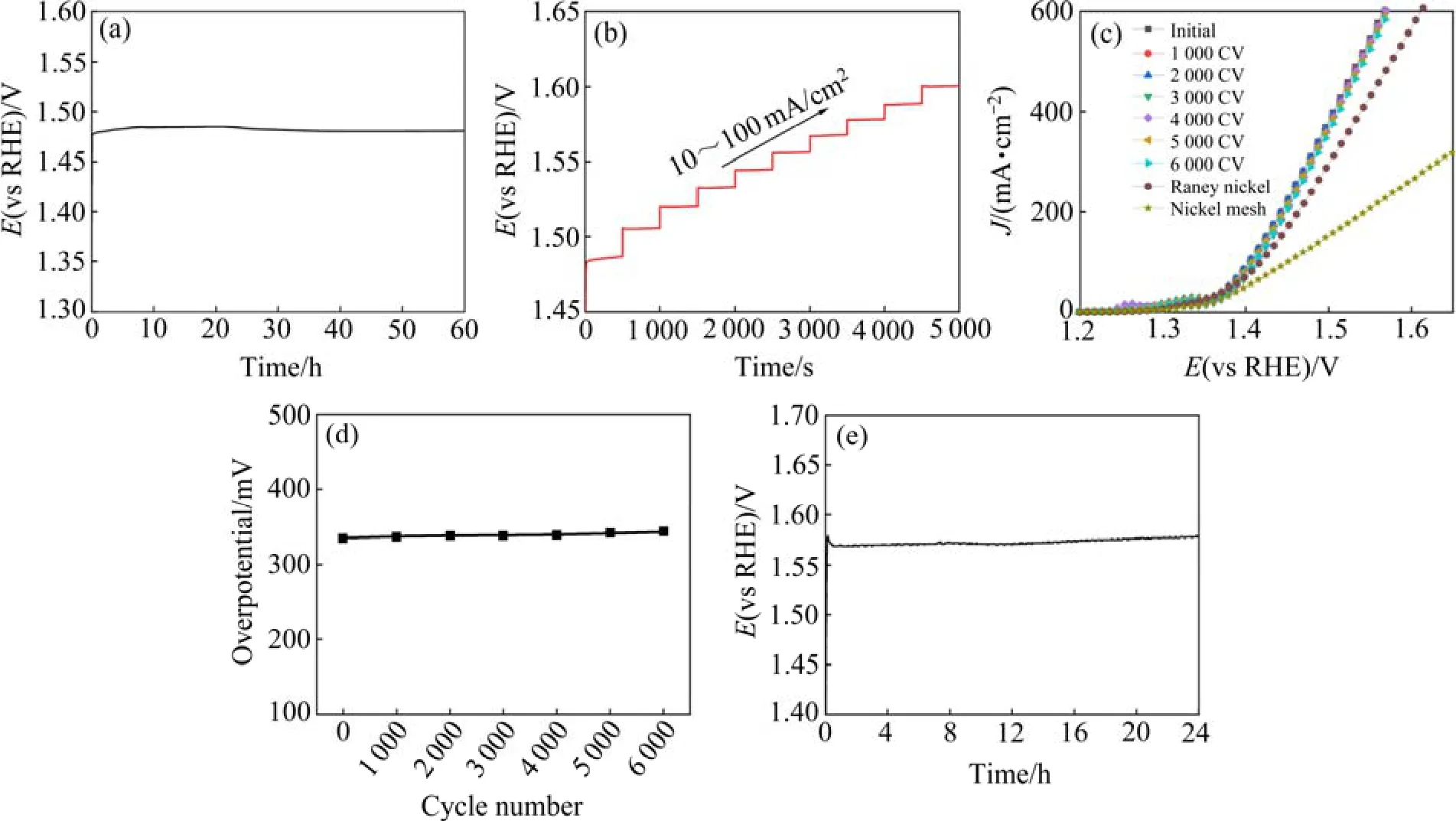
图7 Ni3Fe/Ni3S2/NM 的稳定性Fig.7 Stability of Ni3Fe/Ni3S2/NM
3 结论
1) Ni3Fe/Ni3S2/NM电极在碱性溶液中表现出优异的析氧催化活性,在25 ℃下10 mA/cm2时的过电位为229 mV,双电层电容为6.13 mF/cm2,电荷转移电阻仅为1.78 Ω。在80 ℃下测得600 mA/cm2时的过电位为335 mV,经过6 000 圈CV 循环后,过电位衰减率仅为2.39%,说明该电极有良好的工业应用潜力。
2) Ni3Fe/Ni3S2/NM电极优异的催化活性归因于引入S 后可增强Ni 与Fe 之间的协同效应,降低氧中间体的吸附能,且Ni3Fe 本身的金属特性降低了电极的电荷转移电阻。Ni3S2活性物质暴露出高折射率的{210}晶面与Ni3Fe 的(111)晶面产生协同效应,提供了更多的活性位点。
