改性二氧化锰催化剂催化氧化甲醛的进展*
2023-11-09丁红蕾潘卫国邱凯娜马骏驰张子沂
宋 杰,丁红蕾,2,3,潘卫国,2,3,邱凯娜,张 凯,马骏驰,张子沂
(1.上海电力大学 能源与机械工程学院,上海 200090;2.上海发电环保工程技术研究中心,上海 201600;3.机械工业清洁发电环保技术重点实验室,上海 200090)
0 引 言
改革开放以来,大量挥发性有机化合物(包括烷烃、烯烃、炔烃、芳香烃和含杂原子化合物)向环境排放[1]。预计到2030年,中国工业VOCs排放量将比2010年增加28.75%[2]。甲醛(HCHO)作为典型的室内污染物,主要来源于各种板材使用的脲醛和酚醛树脂胶粘剂[3]。尽管在发达国家,脲醛树脂已逐渐被更环保的材料所取代,但是在发展中或欠发达国家,由于其低成本和有效性,仍被广泛使用。早在2004年,HCHO就被国际癌症研究机构(IARC)确认为1级致癌物[4],长期吸入会对人体造成严重破坏,引起神经系统功能障碍和呼吸系统疾病[5,6]。此外HCHO极易与其他空气污染物发生二次反应,产生具有刺激性气味和慢性毒性的气体,从而产生光化学烟雾和二次污染物[7]。如表1所示,研究人员开发了许多空气净化技术,以减少甚至消除HCHO的产生和排放。其中,热催化氧化技术能够在较低温度下将HCHO转化为无害的CO2和H2O,具有效率高、成本低、无有毒副产物、对浓度要求低等优点[8]。
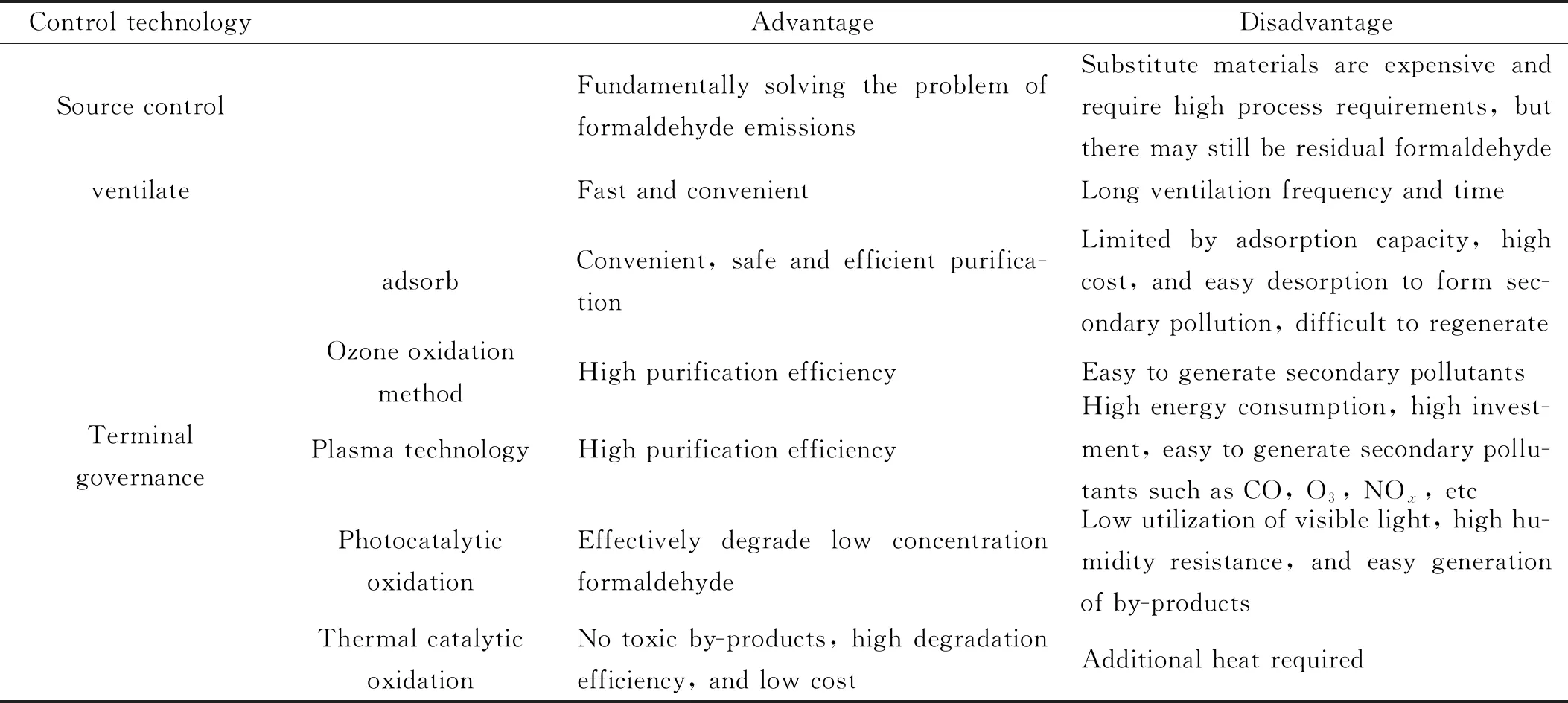
表1 不同控制策略的优缺点Table 1 Advantages and disadvantages of different control strategies
用于HCHO处理的催化剂一般分为两类:贵金属基催化剂(Pt、Pd、Au等)和非贵金属基催化剂(Mn、Cu、Co、Ce等)。贵金属催化剂虽然在室温下对HCHO具有优异的催化性能[9],但受限于其高昂的成本,易中毒倾向和热稳定性差等问题,大规模工业化应用很难开展。近年来,研究人员更加注重开发相对廉价的低温催化氧化材料。MnO2由于独特的储氧能力、电子传递特性以及储量丰富、晶相结构易调节[10]等特点,成为当前研究最为广泛的非贵过渡金属氧化物之一。Sekine等首先比较了多种金属氧化物对HCHO的去除率。其中MnO2表现最好,在室温下,24 h内HCHO去除率可达91%。与其他过渡金属氧化物相比,MnO2的低温催化活性甚至与部分贵金属催化剂相当[11],并且对于混合VOCs也具有较好的催化氧化性能[12]。本文首先介绍了MnO2材料的类型和基本结构,然后综述了近年来MnO2基催化剂在HCHO氧化领域的研究进展,详细阐述了近年来在晶面结构、表面形貌、空位缺陷、与其他材料复合等方面取得的成果,最后简要叙述了催化机理。希望本文对新型二氧化锰基材料的设计和合成提供一定的参考价值。
1 MnO2结构
MnO2是一种结构复杂的氧化物,其基本构建单元是[MnO6]的规则八面体,其中Mn和O原子分别位于八面体的中心和8个顶点[13]。[MnO6]八面体通过不同的堆积方式形成链状结构,之后相互勾连形成隧道或层状结构。其中α- MnO2、β- MnO2、γ-MnO2呈隧道结构;δ-MnO2呈层状结构。图 1为几种常见MnO2晶型的结构示意图。表2为不同晶型MnO2的晶胞参数数据。
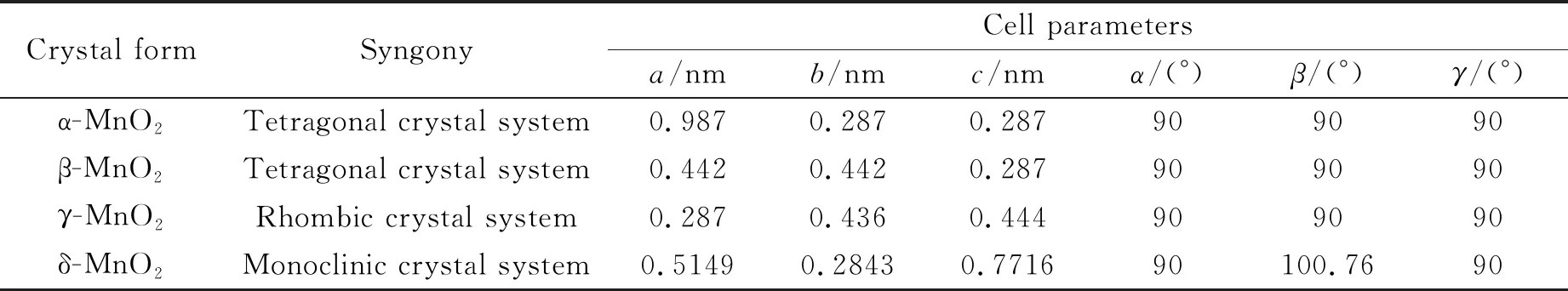
表2 各种MnO2晶胞参数Table 2 Various MnO2 unit cell parameters
1.1 隧道结构
MnO2框架由[MnO6]八面体作为基础构建单元,这些[MnO6]八面体共享角或边,形成链状板,并进一步交叉连接,堆叠,形成各种不同的隧道结构。如图1所示,其中α-MnO2由[1 × 1](尺寸为0.19 nm)和[2 × 2](尺寸为0.46 nm)通道组成,β- MnO2由[1 × 1]通道组成,γ- MnO2由[1×1]和[1×2](尺寸为0.23 nm)组成[14]。这种隧道结构通常含有K+和Na+等离子,以保持结构稳定和电荷平衡。
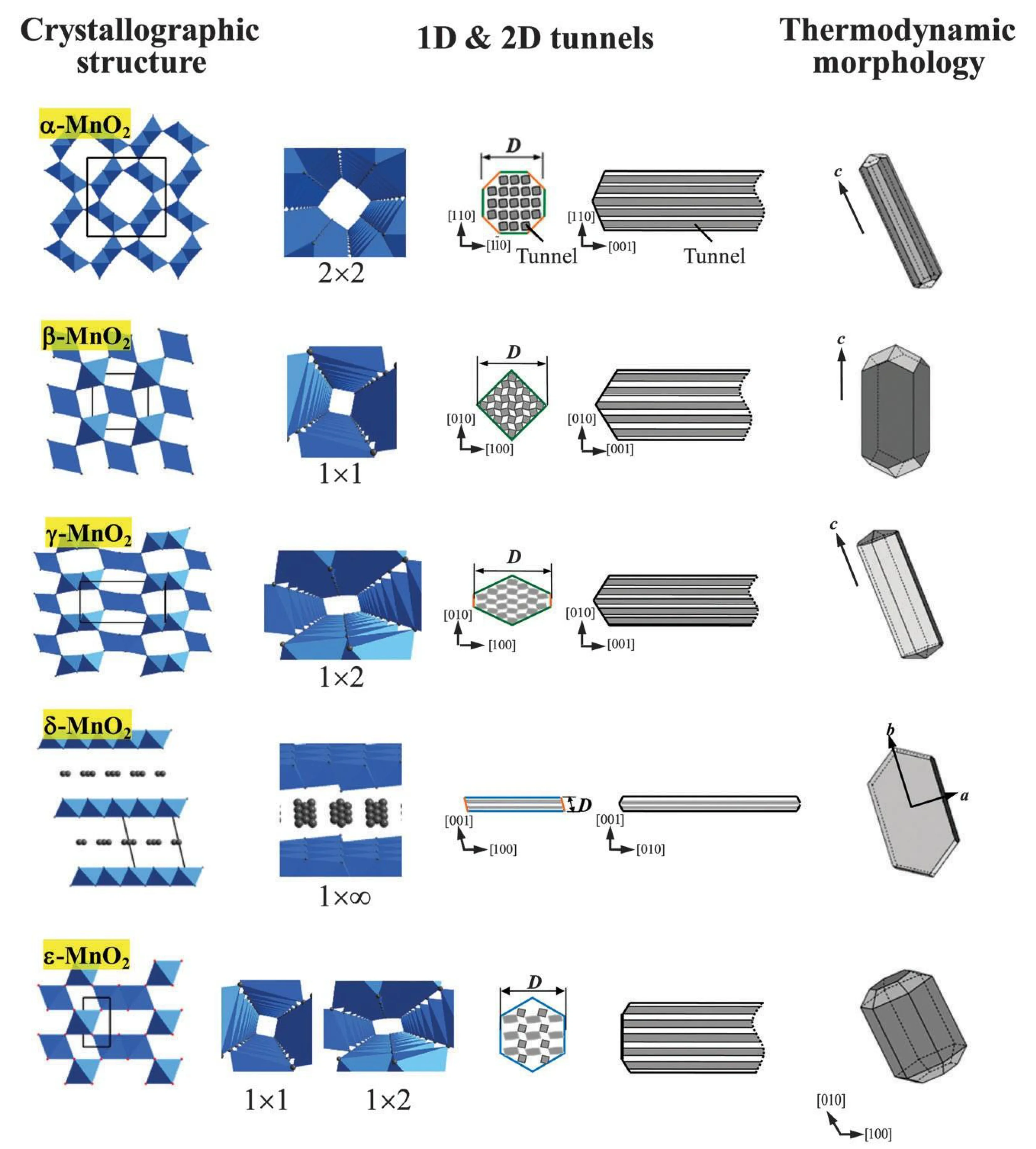
图1 不同晶体结构的MnO2示意图[13]Fig.1 Schematic diagram of MnO2 with different crystal structures[13]
Chen等[15]比较了3种隧道结构MnO2,即软锰矿(β-MnO2,1 × 1隧道)、隐钾锰矿(α-MnO2,2 × 2隧道)和钡镁锰矿 (3 × 3隧道)对HCHO的氧化活性。如图2所示,α-MnO2具有与HCHO气体分子大小相似的2×2隧道结构,对HCHO具有较强的亲和力,这可以加强HCHO分子在MnO2孔隙中的内部扩散,加速其转化为CO2和H2O,从而在 140 ℃时实现HCHO的完全转化。Bai等[16]以KIT-6为硬模板,通过三维介孔合成β-MnO2。在相同条件下,3D-MnO2与原有β-MnO2的1维隧道结构相比,具有更大的介孔结构和更高的比表面积,有利于催化反应中反应物和产物的传质扩散。3D-MnO2可以在130 ℃完成100%HCHO的转化,远高于β-MnO2(180 ℃)。
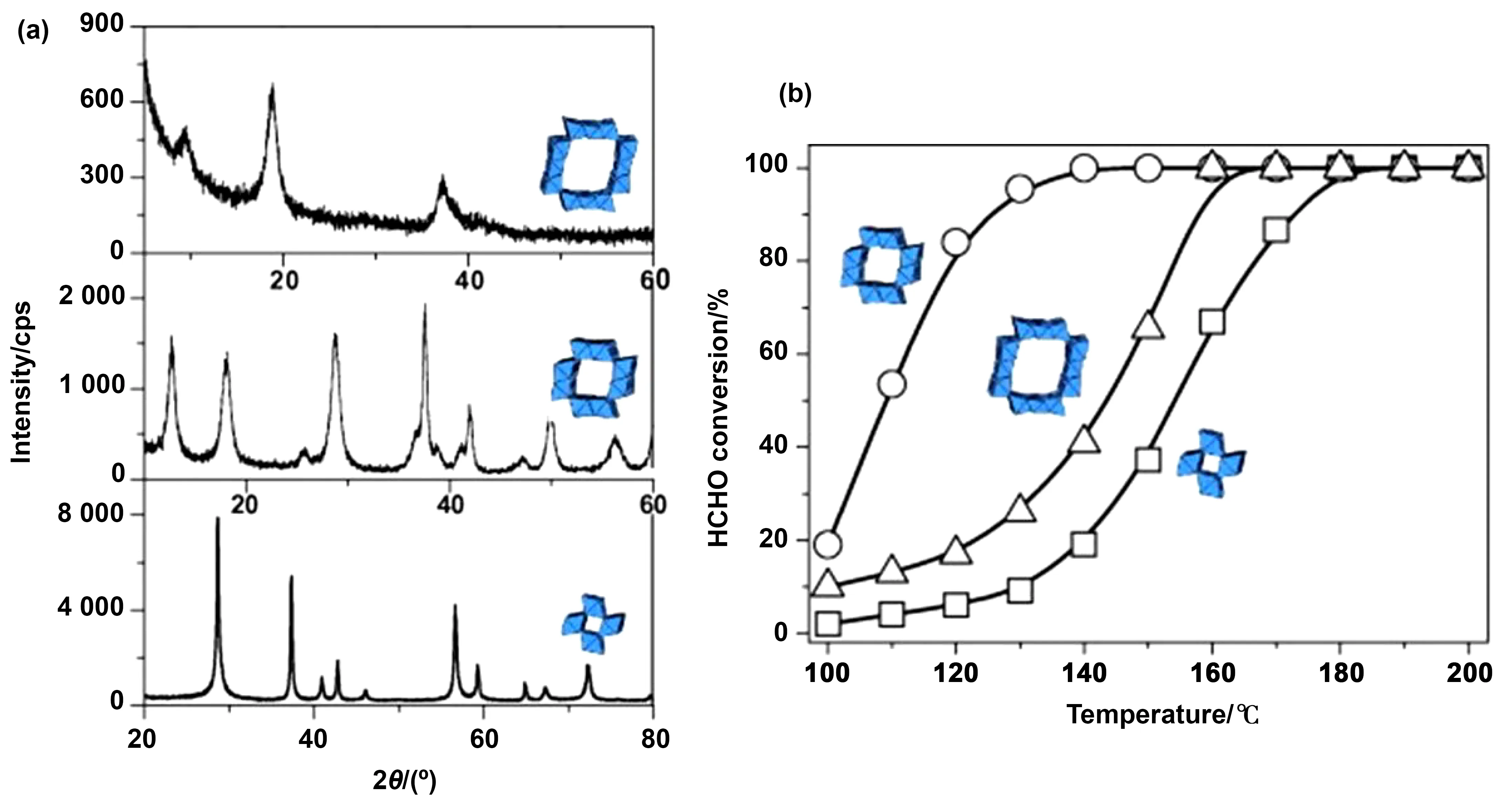
图2 (a)3种不同方形隧道尺寸锰氧化物的晶体结构模型;(b)1×1,2×2和3×3的隧道效应;(c)隧道锰氧化物催化剂对HCHO催化性能[15]Fig.2 (a) Crystal structure model of manganese oxide in three different square tunnel sizes;(b) tunneling effect of 1×1,2×2 and 3×3;(c) catalytic performance of tunnel manganese oxide catalyst for HCHO[15]
1.2 层状结构
二维层状材料因其大比表面积、优异的电隔离性、高导热性和化学稳定性而引起了研究人员的广泛兴趣[13]。δ-MnO2通过共享[MnO6]八面体的边缘形成超薄的2d层状结构。由于超薄纳米片的电子约束,它表现出独特的电子和物理化学性质[17]。据报道,δ-MnO2的催化活性更多依赖于其表面还原反应(空位缺陷和氧物种等),而非形态结构、粒径或表面积[18]。与α-MnO2相比,具有层状结构的δ-MnO2由于其特殊的电子性质,在反应性能上更具竞争力。
Wang等[19]比较了隧道结构(α- MnO2、β- MnO2和γ-MnO2)和单层结构(δ-MnO2)对HCHO的的催化活性。结果表明,δ- MnO2对HCHO表现出最高的催化性能。在δ- MnO2纳米片上存在大量的Mn和O空位,能改变周围元素的电荷密度分布,使其具有良好的电子迁移率。同时分层结构增加了HCHO的吸收和扩散速度,并提供了更多的反应位点,在80 ℃实现了100%的HCHO转化率。Zhang等[20]研究了α- MnO2、β- MnO2、γ- MnO2和δ- MnO2的吸附氧和晶格氧等活性氧在室温下对HCHO催化活性的影响。一般来说,MnO2表面的吸附氧在反应中转化为羟基自由基等氧自由基,并附着在MnO2的隧道/夹层上。结果表明,δ- MnO2在低温条件下对HCHO具有较好的催化性能。这主要是因为δ- MnO2中晶格氧的比例较大,有利于 HCHO在催化剂表面的吸附,促进了中间产物的转化和最终产物的解吸。
综上所述,具有隧道结构的MnO2使HCHO更容易通过外扩散和内扩散进入催化剂,与催化剂充分接触,获得优越的催化活性;而层状结构的MnO2可能具有缺陷和空位等电子性质,宽阔的层间结构(0.7 nm)可以接受比隧道结构更大的分子(如H2O),层间由H2O分子转化而来的氧自由基对反应物分子具有较强的物理或化学吸附性能,从而使HCHO更容易与催化剂表面的反应活性位点接触,获得更好的反应性能。
2 MnO2催化剂催化氧化HCHO
MnO2作为降解HCHO的主要催化剂之一,提升其催化性能对室内环境和人体健康有着重要意义。下文主要介绍了调整晶型形态、暴露特定晶面、控制表面形貌、制造空位和缺陷、与其他非金属材料复合等技术手段以加强MnO2对HCHO的催化性能。
2.1 各种晶体形态
不同晶体形态的MnO2在反应性能存在差异。研究表明,不同晶型催化活性的差异主要来源于表面活性氧的种类和占比。Chen等[21]合成了具有α-、β-、γ-和δ-4种晶相的MnO2催化剂。MnO2独特的Mn-O键和不同晶相的还原性赋予了MnO23种氧,即弱结合氧、表面晶格氧和晶格氧。δ-MnO2中弱结合氧的存在使其对HCHO的催化性能最好,在100 ℃时达到100%的去除率(如图3)。类似的Zhang等[20]通过水热法制备了α-、β-、γ-和δ-MnO2催化剂,测试其对HCHO的催化氧化能力,得出了相同的实验结果:δ-MnO2在80 ℃时能完全转化HCHO,而α-,β-,γ-MnO2则分别需要125,200和150 ℃。进一步研究发现δ-MnO2呈直径为2 ~3 μm的球形形貌,由许多交错的纳米薄片组成。层状结构能产生更多的活性晶格氧,这也是δ-MnO2具有良好性能的主要原因。
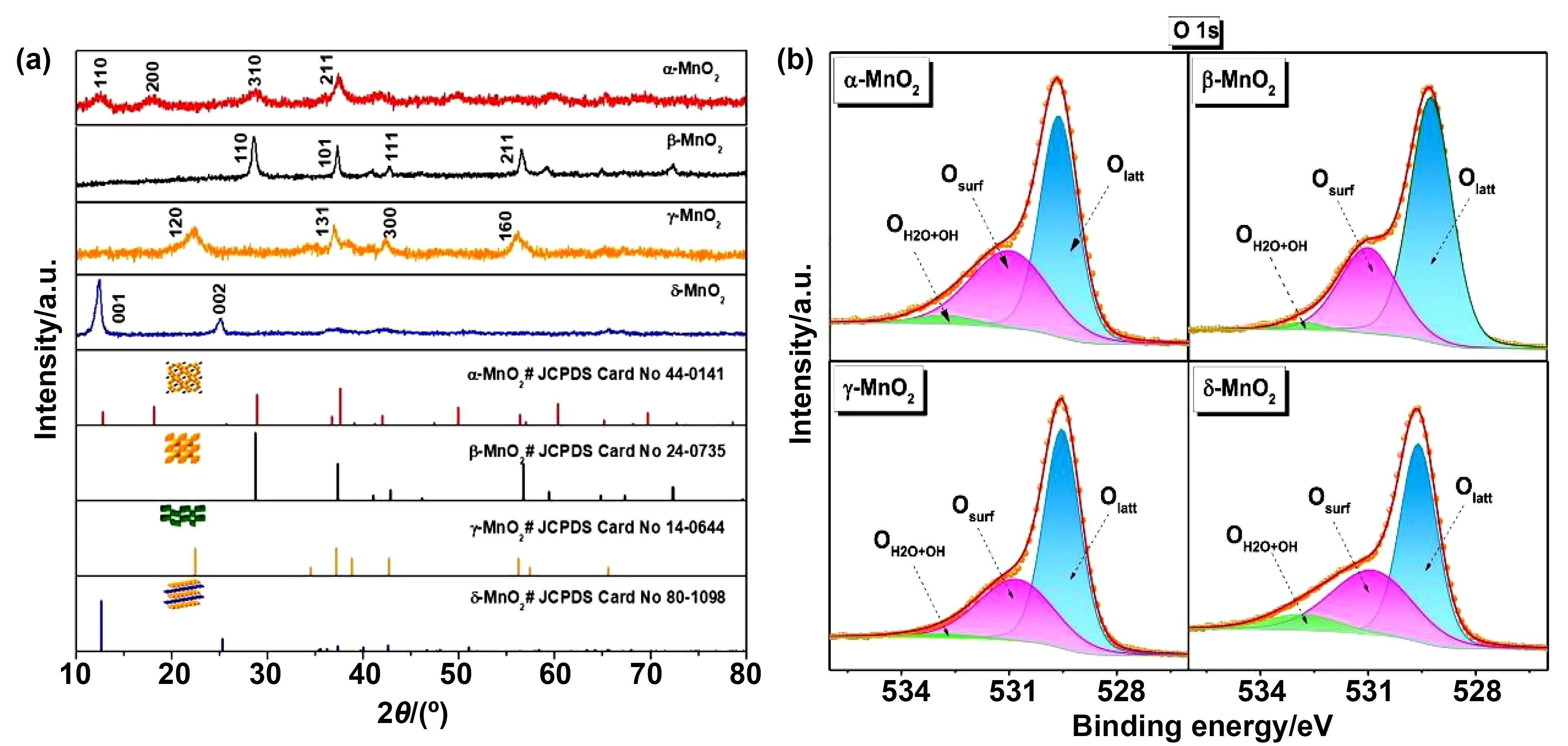
图3 (a) MnO2的XRD谱图;(b)不同晶相MnO2的O1s XPS谱。[21]Fig.3 (a) XRD spectra of MnO2;(b) O1s XPS spectra of different crystal phases MnO2[21]
2.2 不同暴露晶面
除晶型外,不同暴露晶面也将对催化性能产生一定的影响。MnO2不同暴露晶面具有不同的暴露原子,扭曲的电子结构和不同的活性表面位点,从而使其表现出不同的物理化学性质。例如Rong等[22]制备了具有不同暴露晶面(100、110和310面)的α-MnO2纳米线(如图4所示)。这些样品均呈矩形截面。由于α-MnO2纳米线通常沿[001]方向(c轴)生长,因此4个侧壁将是主要的暴露面。其中α-MnO2-[310]具有最高的表面能,能够促进O2和H2O的吸附活化,增强α-MnO2在HCHO上的吸附。因此,α-MnO2-310在室温下对HCHO的催化性能最好。通过调节反应条件,如K+浓度、反应温度、pH等,可以有效地控制MnO2不同晶面的暴露。这对指导二氧化锰催化剂的制备和研究其催化氧化机理具有重要意义。
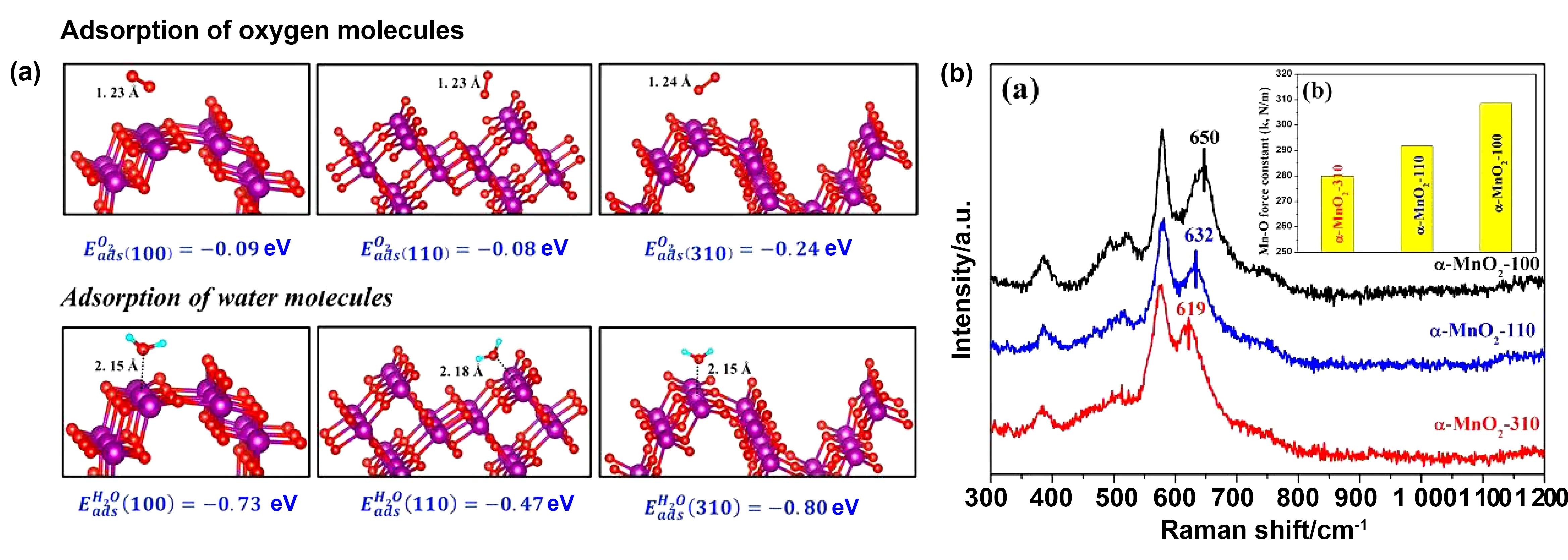
图4 (a)不同暴露面α-MnO2对O2和H2O的吸附能。红色小球是氧气。青色小球是氢,紫色大球是锰。(b)不同暴露面α-MnO2的拉曼光谱和(c)不同暴露面α-MnO2对应的Mn-O键力常数[22]Fig.4 (a) Adsorption energy of O2 and H2O α-MnO2 at different exposed surfaces.The red globule is oxygen.The cyan globules are hydrogen,and the purple globules are manganese;(b) Raman spectra of different exposure surfaces α-MnO2 and (c) Mn-O bond constants corresponding to different exposure surfaces
2.3 表面形貌
气相反应物的吸附和反应主要发生在MnO2的表面和孔内,因此可以通过加强外扩散或内扩散能力来提高MnO2的催化活性。通过对其形态结构和多孔结构进行设计和优化,可以有效改善反应气体在MnO2上的扩散与反应。
一般来说,表面形貌与材料的比表面积密切相关。比表面积越高,其外部扩散性能越好。例如Miao等[23]发现采用插层剥离策略将纳米片剥离成单层状MnO2后,比表面积由101.3 m2/g扩大到234.1 m2/g,提供了更多的附着面积,使HCHO更容易通过单层MnO2纳米片,大大增强了HCHO在室温下的扩散能力和催化效率。常见的MnO2形貌有纳米纤维、纳米片、三维网状结构、纳米棒、纳米球。其中纳米纤维、纳米片和三维网格结构具有简单的形态结构,由于其比表面积小,只有在较低的气体流速和相对压力下才能保证气体与活性位点的充分接触;纳米棒和纳米球一般是由纳米纤维、纳米片和三维网格结构膨胀或破碎而形成的复杂结构。它们通常具有较大的比表面积,HCHO更容易被其吸附。此外,交错的层结构可以延缓气流速度,能保证在较高的流量或相对压力下反应物和活性位点的充分接触。Su等[24]合成了宽高比为30~100 nm的α-MnO2纳米棒。研究表明,当反应物扩散到催化剂上时,交错层结构可以减缓气流速度,使反应物与活性部位充分接触,在50 ℃下,在浓度为15×10-6时,HCHO去除率达到62%。Opembe等[25]采用微波辅助法合成了纳米纤维团聚形成的纳米球,纤维直径更窄,比表面积更大(约153 ~213 m2/g),是常规方法合成的α-MnO2的2倍。后续的活性测试也表明其对HCHO极佳的催化活性。Rong等[26]通过冰模板法挤压冰晶,使MnO2颗粒之间的范德华力显著增强,建立了3D-MnO2骨架(图5)。因为其高孔隙率的三维结构可以显著增强3D-MnO2对HCHO的扩散性能。实验结果表明,3D-MnO2具有良好的传质性能,不仅促进了HCHO向3D-MnO2的内部扩散,而且使CO2能够快速分离,从而减少了中间组分的积累。此外,纳米颗粒表面没有残留有机溶剂,使得HCHO更容易与3D-MnO2表面的活性位点结合。一般来说,HCHO对不同形态结构MnO2的外扩散性能依次为纳米球>纳米棒>三维网格结构>纳米片>纳米纤维。未来可以考虑通过优化形态结构来提升MnO2的活性。

图5 (a)3D-MnO2制备流程;(b) 冷冻过程的冰晶生长示意图;(c)KMnO4与D-(+)-葡萄糖之间的氧化还原反应过程;;(d)3D-MnO2 立于狗尾草上的照片[26]Fig.5 (a) 3D-MnO2 preparation process;(b) schematic diagram of ice crystal growth during the freezing process;(c) the redox reaction process between KMnO4 and D-(+)-glucose;(d) photo of 3D-MnO2 standing on a dog’s tail[26]
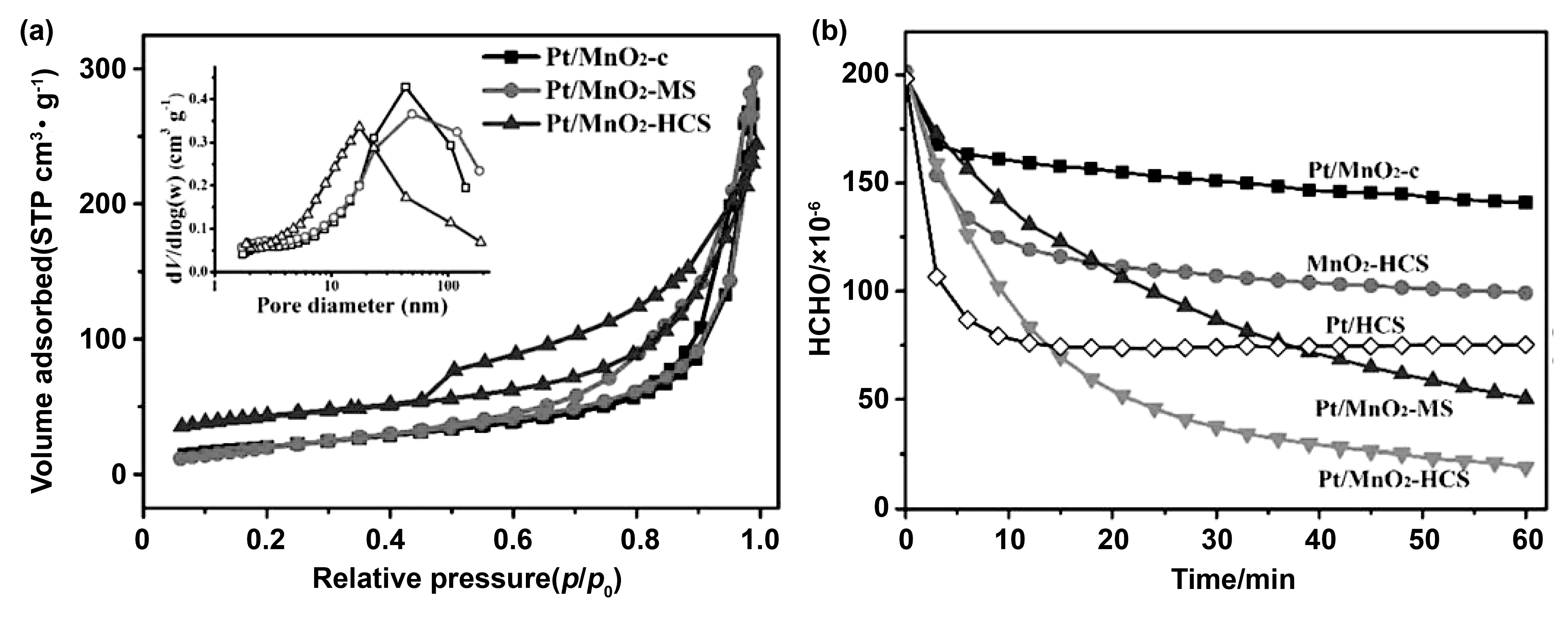
图6 (a)样品的氮吸附-解吸等温线和相应的孔径分布曲线;(b)HCHO转化率[27]Fig.6 (a) Nitrogen adsorption-desorption isotherm of the sample and corresponding pore size distribution curve;(b) HCHO conversion rate[27]
在内扩散过程中,孔隙体积(Vp)、孔径(Dp)等参数与材料的内扩散性能密切相关。在合成催化剂时,孔隙体积和尺寸越大的催化材料具有更好的内部扩散性能。一般来说,多孔材料按照孔径大小分为微孔(<2 nm)、中孔(2 - 50 nm)和大孔(>50 nm)。Sun等[27]发现MnO2的不同孔径分布在一定程度上与其催化性能有关。他们制备的3种催化剂均呈现介孔结构,其中Pt/MnO2-HCS主要孔隙为中孔(<40 nm),而Pt/MnO2- MS和Pt/MnO2以大孔为主。这种多孔结构可以减少空气阻力,促进气态反应物在催化剂上的有效扩散,但后者的大孔结构能极大地促进气体反应物向内部活性位的扩散,从而导致催化活性的增强。Cao等[28]制备的高孔隙率MnO2催化剂可以减小气流阻力,促进气体内部扩散。在反应物分子向活性位点扩散过程中,独特的孔网络极大地促进了扩散速率,有利于后续反应的进行。Chen等[29]发现催化膜微反应器(CMMR)具有分散良好的膜孔,可以防止固定在膜孔中的MnO2/ZIF-8团聚,加强HCHO与MnO2的接触,使HCHO更高效地向内部扩散。CMMR的膜孔可以将浓度边界控制到亚微米级,从而缩短传质距离,显著提高扩散效率。将MnO2/ZIF-8加载到CMMR上后,MnO2/ZIF-8 CMMR的比表面积增加到339 m2/g。此外,在CMMR膜孔中填充MnO2可形成孔径为大于10 nm的微孔。HCHO去除率也从32.6% (MnO2/ZIF-8)增加到63.4% (MnO2/ZIF-8 CMMR)。
2.4 氧空位
长期以来,人们普遍认为多相催化剂的活性中心与其表面缺陷密切相关[19]。当MnO2中形成一个氧空位时,就会产生两个多余的电子。因此,氧空位是一个富电子中心。如果H2O/O2被吸附在氧空位附近,相应的电子会发生转移,产生大量的表面活性氧,促进其表面吸附性能和反应性的增强[30]。例如Zhu等[31]利用草酸铵抑制了δ-MnO2晶体的生长,导致结晶度差,形成比表面积较大的纳米球(由δ-MnO2纳米片组成),提高了表面氧和晶格氧的还原性和迁移率,产生了更多的氧空位,从而提高了反应性能。
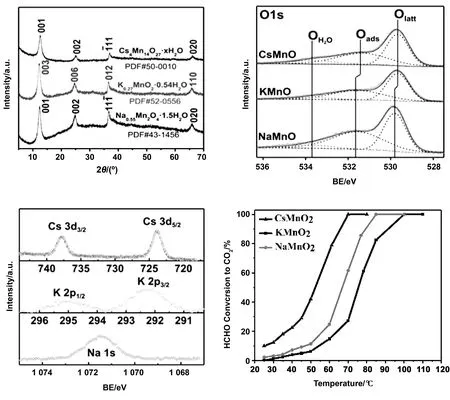
图7 (a) XRD,(b) O1s的XPS,(c)阳离子(Na,K,或Cs)的XPS,(d) HCHO氧化的活性图谱[34]Fig.7 (a) XRD;(b) XPS for O1s;(c) XPS for cations (Na,K,or Cs);(d) activity profile of HCHO oxidation[34]
此外,研究发现,过渡金属离子的引入可以带来更强的吸附/解吸能力、优良的界面结构和更高的表面反应活性。通过取代锰离子,一些金属阳离子(如Cu2+、Ce2+或Co2+)被成功地纳入MnO2骨架中。Gao等[38]研究了过渡金属离子掺杂对提高α-MnO2纳米线反应性的影响,活性顺序依次为Cu0.1MnOx>Co0.1MnOx>Ni0.1MnOx>Fe0.1MnOx>MnO2。DFT计算指出Cu掺杂MnO2更容易形成氧空位,而氧空位是影响催化反应速率的关键。Zhu等[31]合成了不同掺杂比例的Ce修饰δ-MnO2(Ce-MnO2)。结果表明,Ce掺杂可以抑制MnO2晶体的生长,在形成更大的比表面的同时促进氧空位的产生。综上所述,金属离子掺杂是人为制备氧空位和表面活性氧的有效手段。
2.5 锰缺陷
阳离子空位作为关键缺陷之一,是调控催化剂表面电子性质、提高反应性能的有效方法[39]。Wang等[40]证明了在超薄MnO2纳米片优异的催化性能来源于表面形成大量空位缺陷。由于锰空位的存在,Olatt周围的电子密度降低,从而改变了锰空位附近晶格氧的结合能和密度。MnO2样品中锰空位含量随Mn3+/Mn4+比值的变化而变化。Liu等[41]首次利用碱金属阳离子Li+、Na+、Rb+和NH4+取代锰离子,形成的掺杂α-MnO2出现了大量锰缺陷,提高了MnO2的热稳定性和活性。研究发现,这些金属离子的掺杂显著提高了MnO2的表面氧活性,从而提高其室温下的HCHO去除率。同时,K+在VMn附近的存在也促进了表面吸附剂的活化。由于VMn周围K+与不饱和氧之间的键比Mn-O键弱,这些不饱和氧更容易移动并作为活性氧增强对HCHO的催化反应活性[42]。类似的Yusuf等[43]将不同含量的K+混合到MnO2中,显著提高了Mn4+和晶格氧的含量。K0.5表现出最佳的催化活性,可在70 ℃完全转化170 ×10-6HCHO。
适量的过渡金属元素与MnOx结合,可以有效抑制MnOx晶体的生长或非晶化,促进锰基固溶体或其他混合氧化物相的形成[44],使MnOx的晶格形变产生大量的锰空位,增强其氧化还原性能[45]。Ce和Mn的相互作用会削弱Ce-O键之间的作用力,使CeO2中的活性氧更容易释放并参与有机污染物的降解[46],实现HCHO低温氧化。Li等[47]开发了一种制备CeO2掺杂δ-MnO2催化剂的方法,其特点是Mn4+/Mn3+摩尔比高,低温还原性好。高度分散的CeO2纳米颗粒与δ-MnO2的协同作用提高了HCHO催化剂的活性和稳定性。Quiroz等[48]对酸处理合成的MnOx-CeO2催化剂进行了研究,发现当摩尔比Mn:Ce >0.5(高于Mn的溶解极限)时,MnOx-CeO2颗粒之间存在Mn2+溶解,使材料具有一定的空隙和较大的比表面积。同时,通过Mn离子之间的歧化反应,可以得到氧化态较高的Mn离子,从而提高对HCHO的催化氧化活性。Co3O4也是一种很好的复合材料,MnO2和Co3O4的混合可以使合成的材料具有更强的氧化还原反应活性。Huang等[49]制备的CoxMn3-xO4(x=0.65)纳米片对HCHO的氧化表现出优异的催化活性(T100=100 ℃)和稳定性(24h内HCHO转化效率下降不到3%)。这是由于Co的加入使催化剂Mn4+含量大幅度增加,同时两者的协同作用也保证了整体稳定性和活性。
2.6 与非金属材料复合
除了上述方法外,一些学者致力于利用非金属材料来使MnO2材料具备更为优异的吸附性能和催化活性。碳基材料因其多孔结构、优异的光学热学性能和独特的表面电子态而被广泛应用于集成复合材料中[50]。Yang等[3]制备了不同负载量的MnO2/AC 催化剂,均可以在室温条件下72 h内完成85%以上HCHO的去除。Li等[51]利用原位还原法将水钠锰矿负载于活性炭颗粒上。后续实验结果表明,在室温下,该复合催化剂可以在1 h内净化70%的HCHO,2.5 h内完成100%去除。其优异的催化性能主要归功于活性炭的多孔结构和优异的吸附性能。此外Lu等[52]发现石墨烯的存在对HCHO的吸附有积极的影响。在低温下(<100 ℃),GMn杂化催化剂的HCHO转化率较高。石墨烯的存在导致表面羟基的数量增加,暴露的Mn4+活性位点数量增加,同时,石墨烯纳米片的强导热性也使其吸收的热量转移到MnO2上,最终变相提高了活性。Liu等[53]合成了纳米碳修饰的MnO2,发现该复合催化剂可以在室温下将低浓度(0.5 mg/m3)的HCHO氧化为CO2(92%)。纳米碳不仅限制了MnO2的生长,导致缺陷位点含量高,还促进了接触界面上的电子转移。Wang等[54]将MnO2分散到碳球表面。C@MnO2催化剂的核壳结构可以在室温下实现低浓度甲醛的100%转化。MnO2与C之间形成的亲密界面促进了吸附和后续的催化反应。此外,碳球的引入也促进了更多的不饱和MnO2位点,这些位点对HCHO氧化十分有利。
考虑到粉末或颗粒状催化剂在实际应用中风阻较大,将MnO2材料负载到纤维素上也是当前很有趣的研究方向。Zhou等[55]将MnO2纳米片原位沉积到多孔纤维素上(8.86%(质量分数) MnO2/纤维素),制备的复合型催化剂催化性能远高于传统MnO2。MnO2的用量和HCHO在纤维素上的吸附量是该复合催化剂活性的主要影响因素。Rong等[56]将超薄纳米片沉积到PET纤维上,该催化剂在室温下即可达到90%以上的HCHO去除率。由于材料失去活性主要是由于中间产物甲酸盐和碳酸盐物种在MnO2/PET表面的积累所致,所以废催化剂只需100 ℃加热即可再生,无需任何漂洗处理,可轻松实现催化材料的有效再生与循环利用。Zeng等[57]利用原位沉积技术成功将MnO2纳米颗粒均匀分散到多孔无纺布纤维上(MnO2/PET),可以在室温下3 h内完成90%以上的甲醛分解,催化性能远高于单一MnO2。
3 MnO2催化甲醛的机理

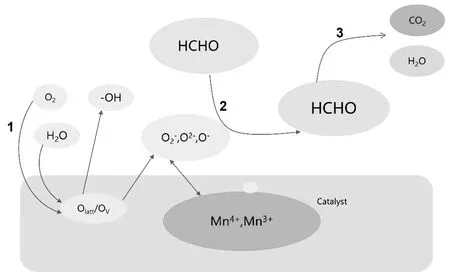
图8 HCHO在MnO2上的MVK机理图Fig.8 MVK mechanism diagram of HCHO on MnO2
研究人员通过原位DRIFTS技术进一步探索了HCHO催化氧化中间体的转化。众所周知,催化剂表面中间体的积累可能导致催化剂失活。对于HCHO的低温催化氧化,催化剂表面碳酸盐的转化和解吸是确保活性位点可用性和进行连续反应的关键步骤[62]。Wang等[63]在之前的研究中报道HCHO的催化氧化通常伴随着表面羟基的消耗。如图10(a)所示,当3D-MnO2暴露于HCHO中时,3 850和1 658 cm-1处的条带强度随着暴露时间的增加而增加。这些带归属于结构羟基(ν(OH))和δ(H-O-H))。由于羟基含量较高,碳酸盐物种更容易被解吸,导致表面碳酸盐物种在其表面的积累较少。3D-MnO2表面羟基能被空气中的水快速补偿。Yusuf等[64]制备了Cu/δ-MnO2,并进行了原位DRIFTS分析。如图10(b)所示,Cu/δ-MnO2上的碳酸盐峰(1 653 cm-1)几乎成为主导峰,这表明随着时间的推移,碳酸盐在表面聚集。研究发现,δ-MnO2中Cu的存在导致碳酸盐的表面堆积,进而导致表面活性位点被覆盖,表面氧的活化和流动也会受到阻碍。

4 结 语
近年来,MnO2催化剂取得了重大突破和进展,使其成为HCHO催化领域最有前途的材料之一。本文从MnO2的结构性质(晶型、晶面、表面形貌、空位缺陷)和与其他材料复合等方面,阐述了近年来MnO2催化氧化HCHO的最新进展。综上所述,未来可以从以下三方面着手提升MnO2的催化活性:MnO2的性能受其形态结构、孔结构的影响较大,纳米球或纳米棒状MnO2能更好地降低扩散阻力,有利于HCHO克服能量势垒,与内表面活性位点接触。介孔和大孔交错间隙的MnO2也能降低扩散阻力,促进HCHO在催化剂结构中的扩散;不同晶体结构和空位缺陷均能调节MnO2催化剂表面的电子性质,有效提高HCHO的吸附/解吸和催化活性,从而加速HCHO的反应;与其他非金属材料复合有利于促进材料的活性成分分散,为催化反应提供空间,解决HCHO在MnO2上扩散和吸附容易饱和的问题,改善MnO2对低浓度甲醛的催化性能。
