BALB/c突变卷毛小鼠基因组学测序差异的分析*
2023-11-02汤紫荣李晓娟姜棋予李兴杰孙慧伟李瑞生
汤紫荣 李晓娟 姜棋予 李兴杰 孙慧伟 李瑞生
(1.中国人民解放军总医院第五医学中心住院与病案管理科,北京 100039) (2.中国人民解放军总医院第五医学中心感染病医学部研究所,北京 100039)
全基因组测序是指应用新型测序仪器测量不同机体基因组间的差异,并通过生物信息手段对与性状关联的遗传变异信息及基因组结构进行分析与注释[1]。测序技术已经由基于双脱氧末端终止法的 Sanger测序技术[2]发展到现在的第二代、第三代测序技术[3],实现了从低读长到超高读长、从光学检测到电子传导检测的双重跨越测序技术,在动物基因组测序发展中起了非常重要的作用[4]。BALB/c突变卷毛鼠是本实验室发现的突变小鼠,经近交培育已成为较为成熟、遗传稳定的近交系突变卷毛小鼠品系[5]。前期对其外观特征以及血液学指标进行了基础性探讨研究[6-7],尤其应用微卫星位点标记对BALB/c突变卷毛鼠进行检测分析,发现该小鼠存在较高的突变率,且与无毛小鼠突变是两个完全不同的突变系[8]。因此本实验利用Illumina PE 150对BALB/c突变卷毛鼠与BALB/c小鼠进行测序,并进行生物信息学分析,以期全面了解突变鼠与BALB/c小鼠之间的基因组差异性,为今后更好地开发和利用该鼠作为优良的动物模型提供可靠的生物学信息基础。
1 材料和方法
1.1 实验动物
正常BALB/c小鼠和BALB/c突变卷毛鼠各1只,SPF级,雌性,6周龄。正常BALB/c小鼠来自突变群体中的正常小鼠,BALB/c卷毛鼠来自于本实验室近交培育,生产许可证号【SCXK(军)2012-0004】。动物饲养在中国人民解放军总医院第五医学中心动物实验中心,使用许可证号【SYXK(军)2017-0016】。本实验通过了动物伦理委员会审查,伦理审批号:IACUC-2017-006。
1.2 实验方法
1.2.1动物组织采集和DNA提取:从BALB/c小鼠和BALB/c突变卷毛鼠后肢分别取一小块肌肉,立即用PBS或0.9%氯化钠溶液漂洗,去除血渍,并剔除结缔组织和脂肪组织,吸干水分;将肌肉块修剪成长宽高均≤0.5 cm的小块(组织块越小,保存效果越好);将处理好的组织样品置于2 mL的旋盖冻存管中,立即液氮速冻,送去北京美吉桑格生物医药科技有限公司。而后对2个样本进行基因组DNA的提取、质检和定量检测,以及后续的建库测序。
1.2.2文库构建及测序:2个样品分别构建1个Illumina PE测序文库,共计2个Illumina 测序文库;插入片段400 bp。Illumina PE 150每个样品提供84 G clean data,Q30值≥80%。
1.2.3信息分析
1.2.3.1原始数据质控和过滤:测序结果得出的原始测序序列(Raw Reads)由于含有带接头的、低质量的Reads,因此为了提高信息分析的质量,必须对Raw Reads过滤,得到Clean Reads,后续分析都在Clean Reads的基础上进行。对下机得到的Raw Data进行质控得到Clean Data。
1.2.3.2数据比对:进行单碱基突变(SNP)和 插入缺失(Indel)的检测及注释;Indel是指基因组中小片段的插入和缺失序列。利用SAMTOOLs检测长度小于50 bp的小片段插入与缺失(Indel)。对结构变异(SV)和拷贝数变异(CNV)进行检测和注释。
2 结果
2.1 基因组基本信息对比结果
本次测序正常小鼠与卷毛小鼠产生的Raw Data分别为122.85 Gb和118.04 Gb,过滤后的Clean Data 分别为119.82 Gb和112.18 Gb,GC含量分别为40.63%和40.48%,表明测序质量合格。GC分布也正常,说明建库测序成功(表1)。正常小鼠与卷毛小鼠的基因组覆盖深度统计(表2)。

表1 测序质量统计表Table 1 Sequencing quality statistics

表2 样品覆盖深度和覆盖度统计Table 2 Sample coverage depth and coverage statistics
2.2 遗传变异检测对比分析
卷毛小鼠的杂合分型SNPs总数显著高于正常小鼠,而其他数值却没有显著差异(表3),同时正常小鼠与卷毛小鼠之间的累计SNPs深度分布也存在显著的差异(图1)。卷毛小鼠与正常小鼠插入缺失(Indels)以及累计Indels深度分布均存在差异(图2,3)。卷毛小鼠与正常小鼠的SV类型中大片段的插入卷毛小鼠大于正常小鼠,而其他类型则小于正常小鼠(表4)。而拷贝数变异(CNVs)结果显示,正常小鼠和卷毛小鼠的拷贝数增加的个数分别为1 270和967个;而拷贝数减少的个数分别为5 027和3 505个。
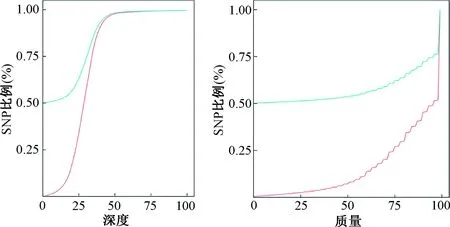
注:红线为卷毛小鼠;蓝线为正常小鼠。Note:The red line is curly mice; The blue line is normal mice.图1 累积SNP深度分布Fig.1 Cumulative SNP depth distribution
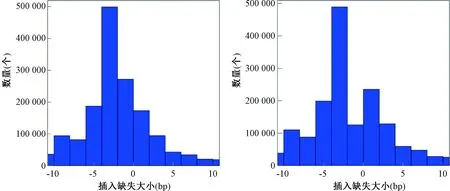
注:A.卷毛小鼠 ;B.正常小鼠。Note:A. Curly mouse; B. Normal mouse.图2 插入缺失(Indel)分布情况Fig.2 Distribution of insertion and deletion
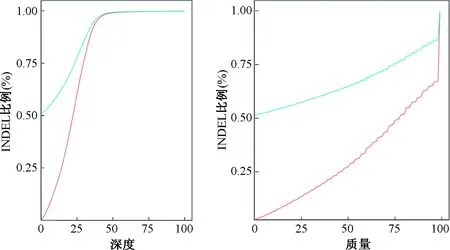
注:红线为卷毛小鼠;蓝线为正常小鼠。Note. The red line is curly mice; The blue line is normal mice.图3 累积Indel深度分布Fig.3 Cumulative Indel depth distribution
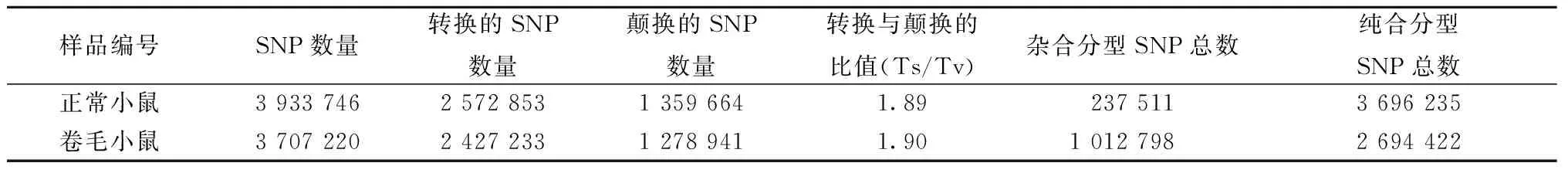
表3 SNP数据统计表(个)Table 3 SNP data statistics(个)

表4 SV预测结果统计表Table 4 Statistical table of SV prediction results
3 讨论
全基因组重测序的特点是具有分析结果快速、准确、灵敏度高和自动化,指在参考已知基因组序列的物种信息基础上,对不同个体整个基因组进行测序,再通过生物信息学分析及在个体或群体水平进行序列差异性分析[9]。那么在测序过程中可以检测到大量与性状关联的遗传变异信息(包括SNP、Indel、SV和CNV甚至新基因等)。研究人员利用全基因组测序方法比较近交系小鼠C57BL/10和C57BL/6的基因组差异,筛选出 4 个小鼠免疫应答差异基因[10]。研究人员完成了美洲大蠊的全基因组测序,也是大蠊属Periplaneta 昆虫的第一个基因组,为美洲大蠊遗传进化分析和药用基因资源挖掘打下了重要基础[11]。但目前对突变小鼠与正常小鼠之间进行对比分析的基因组测序报道甚少。
因此本实验对实验室单独发现的BALB/c突变卷毛鼠与群体中未发生突变的正常BALB/c小鼠进行测序,并进行了生物信息学分析,结果显示:正常小鼠与卷毛小鼠产生的Raw Data分别为122.85 Gb和118.04 Gb,过滤后的Clean Data分别为119.82 Gb和112.18 Gb,GC含量分别为40.63%和40.48%,GC分布正常,测序建库成功。自人类基因组计划完成以来,获得高质量的序列图谱成为了不同物种进行功能基因研究的基础[12]。而遗传变异统计结果显示:卷毛小鼠的杂合分型SNPs总数显著高于正常小鼠,同时正常小鼠与卷毛小鼠之间累计SNPs深度分布也存在显著的差异。卷毛小鼠与正常小鼠插入缺失(Indels)以及累计Indels深度分布均存在差异。卷毛小鼠与正常小鼠的SV类型中大片段的插入卷毛小鼠大于正常小鼠,而其他类型则小于正常小鼠。而拷贝数变异(CNVs)结果显示,正常小鼠和卷毛小鼠的拷贝数增加的个数分别为1 270和967个;而拷贝数减少的个数分别为5 027和3 505个,卷毛小鼠均小于正常小鼠。遗传变异检测分析结果显示SNPs、Indels、SV和CNVs在两种小鼠之间均存在差异,不仅证明了两种小鼠存在遗传差异,且也极大的丰富了遗传资源多样性的研究内容[13]。那么在全基因组水平上,利用SNPs等分子遗传标记存在的差异,也能够较全面解析物种受到的自然选择和人工选择导致的遗传变化[14]。本实验对两种小鼠进行了初步的基因差异对比,但又考虑到即使正常繁育SNP等位点也会发生变异,因此下一步我们将借助一些基因分析软件[15]对新发现的遗传变异差异进行详细的注释,对差异表达基因的功能进行分类,期待能够挖掘这些变异对某些分子和细胞事件有所指示。
综上所述,本研究对BALB/c突变卷毛鼠与同种群中正常BALB/c小鼠进行了全基因组测序,并且发现二者之间存在遗传变异差异,进一步验证了前期实验中两种小鼠各项指标的差异存在。众所周知,全基因组测序可以全面、快速、准确地解析不同品种的分子遗传特征,为品种的不断选育提高及开发利用奠定坚实的基础[16]。我们将对其基因的变异情况进行深入分析,以期更好地开发和应用该突变小鼠。
