电离辐射在肿瘤放射治疗中的应用与放射生物学效应研究进展
2023-10-17冯亚辉涂文玲余道江张舒羽
冯亚辉,涂文玲,余道江,张舒羽
(1.成都医学院第二附属医院 核工业四一六医院,成都 610051;2.国家卫健委核技术医学转化重点实验室,绵阳 621000)
核技术在国防、工业、农业、通讯、交通、环保和资源开发等各个领域都有着广泛的应用,而在核医学诊断、治疗和生命科学研究等多个方面也都发挥着重要作用[1-4]。当用于治疗,尤其是在治疗癌症时,利用电离辐射的主要目标是尽可能精准地照射肿瘤细胞,同时确保健康组织吸收剂量尽可能低。为了实现这一目标,准确判定电离辐射与生物物质相互作用的效应至关重要,了解与总结放射生物学效应对高效、安全利用核技术具有重要的指导作用。
放射治疗(radiotherapy,以下简称为放疗)广泛地应用于临床恶性肿瘤的治疗中。放射治疗始于19世纪末,到现在已有一百多年的历史。1896年,在伦琴发现X射线6个月后,Despeignes在里昂首次利用X射线对癌症患者进行了实验治疗,1898年,居里夫妇发现放射性元素镭后,也很快被许多学者用于肿瘤的内照射治疗[5]。如今,放疗已经是恶性肿瘤临床治疗的三大手段(手术、放疗、化疗)之一,它是利用各种不同种类射线的电离辐射作用杀死癌细胞而治疗肿瘤的无创、局部治疗方式[6]。用于放疗的射线由放射性同位素或放疗设备产生,各种不同种类的放疗设备可产生包括γ射线、X射线、电子束、质子束、中子束、π-介子束、重粒子束等不同种类的射线[7-8]。随着放疗经验的积累和放疗设备的不断改进,目前放疗能够用于治疗绝大部分类型的肿瘤,超过50%的肿瘤患者在治疗过程中需要接受至少一次放疗,肿瘤的放疗使得超过50%的肿瘤患者症状得到缓解或治愈[9-10]。因此,深入了解放疗的基本原理对有效利用各种射线的电离辐射具有重要意义。
1 临床放疗的发展
肿瘤临床放疗的发展与放射物理学和放射生物学的发展密切相关,放射生物学的研究通过揭示放疗中各种射线与机体发生物理反应和生物效应的关系来推动放疗技术的不断进步。传能线密度(linear energy transfer, LET)与相对生物学效应(relative biological effectiveness, RBE)是放射物理学和放射生物学中决定肿瘤放疗效果的重要因素。LET是指电离辐射粒子在其单位长度径迹上消耗的平均能量,X射线、γ射线和中子虽不是直接电离辐射粒子,但它们在与物质发生相互作用后会产生次级带电粒子,因此LET的概念同样适用[11]。RBE是一个相对的概念,最开始以最先被发现的X射线的生物效应为基准,后来建议采用γ射线为标准,因此RBE通常被定义为X射线或γ射线引起某一生物效应所需吸收剂量与所观察的电离辐射引起相同生物效应所需吸收剂量的比值,即为该种电离辐射的相对生物效应[12]。RBE与LET通常呈现正相关关系,在吸收剂量相等的情况下,不同种类的电离辐射所产生的生物效应不同,通常高LET射线的生物效应大于低LET的生物效应。肿瘤放疗中常用的X射线、γ射线和电子束等都为低LET射线,而质子、重离子、中子、α粒子等射线的LET相对较高,因此相同条件下高LET粒子所造成的放射生物学效应比较强。对肿瘤临床放疗来说,放射物理学和生物学研究结果最终应服务于临床放疗,提高肿瘤放疗的疗效。
在过去的一个世纪里,放疗技术的不断进步为临床的放疗实践提供了强大的动力,而且逐渐改变着肿瘤临床治疗的指南。上世纪五六十年代广泛使用的放疗设备60Co治疗机现已逐步退役并被直线加速器取代[13]。加速器能够通过电磁场将带电粒子加速直接得到电子束、质子束或重离子束,由被加速的粒子轰击不同的靶材又会产生X射线、中子束、γ射线、π介子等多种类型的粒子束。多种基于高LET粒子的新疗法正在用于癌症治疗[14-17]。比如具有高LET特性的质子和重离子,其射线的能量沉积在射程末端急剧升高,形成尖锐的布拉格(Bragg)峰,这些独特的物理性质使其能量能够在射程末端的肿瘤部位大量沉积,引发较高的RBE,已经逐渐被用于肿瘤的临床放疗中[18-19]。在放疗的过程中,不可避免地会对正常的组织进行照射,这会对正常的组织产生各种不良反应。近年来,三维适形放疗(three-dimensional conformal radiation therapy, 3D-CRT)技术和调强适形放疗(intensity modulated radiationtherapy, IMRT)技术的快速发展为有效降低放疗对正常组织的辐射损伤做出了重要的贡献[20]。立体定向放射治疗技术(stereotactic body radiation therapy, SBRT)则利用每次高剂量的放疗,通过少数几次分割照射即能达到根治性剂量,消灭肿瘤,SBRT在多种早期肿瘤的治疗中取得很好的疗效[21]。这些不同种类放疗技术的快速发展为满足复杂的肿瘤治疗需求提供了无限可能。
总之,肿瘤临床放疗的发展离不开放射物理学和放射生物学的研究与发展。鉴于高LET粒子具有的高RBE放射生物学特性,它们的广泛应用可能将临床放疗从常规辐射带入到粒子辐射的时代。随着放疗技术和计算机技术的不断发展,临床放疗必将向精细化和个体化发展,肿瘤放疗的控制效率和并发症的发生问题都将大大改善。
2 电离辐射与DNA损伤反应
肿瘤临床放疗的发展离不开肿瘤放射生物学的深入研究,准确判断电离辐射与生物物质相互作用的效应,明确电离辐射所产生的生物学效应对提高肿瘤放疗疗效至关重要。肿瘤放疗的目的是阻止肿瘤细胞增殖并诱导肿瘤细胞死亡,但是放疗通常不会立即杀死细胞,肿瘤细胞死亡可能在放疗后持续几天甚至几个月[22-23]。当电离辐射入射到人体组织或器官时,它可以直接作用于人体组织或器官的细胞分子,损伤DNA,它还能电离细胞内的水产生活性氧(reactive oxygen species, ROS)造成间接的DNA损伤[24-25]。由于肿瘤细胞的高复制率和DNA损伤反应(DNA damage response, DDR)途径的缺陷,肿瘤细胞比正常细胞更容易受到电离辐射的影响[26-27]。电离辐射诱导的DNA损伤示意图示于图1。DNA是电离辐射作用最主要的靶分子,暴露于电离辐射中的细胞,其基因组DNA能被射线直接或间接破坏而产生损伤,产生单链断裂(single strand breaks, SSBs)、双链断裂(double strand breaks, DSBs)和DNA簇损伤(clustered DNA Lesions, CLs)[28-30]。细胞可以通过DDR对基因组DNA中的损伤做出反应,特别是DSBs,这种反应是在DNA损伤的几分钟内通过复杂的DDR网络对受损DNA进行反应而启动[31]。DNA受到辐射损伤后,能够通过非同源末端连接(non-homologous End Joining, NHEJ)或同源重组(Homologous Recombination, HR)等途径进行自我修复,而错误的修复会造成双着丝粒、无着丝粒、染色体易位、着丝粒环、染色体断片等染色体畸变[32]。DNA损伤是电离辐射造成一系列细胞和组织反应的最主要和内在的因素,DDR信号通路是辐射暴露后影响细胞周期和细胞命运(死亡或存活)最关键的操纵因素[25,31-33]。
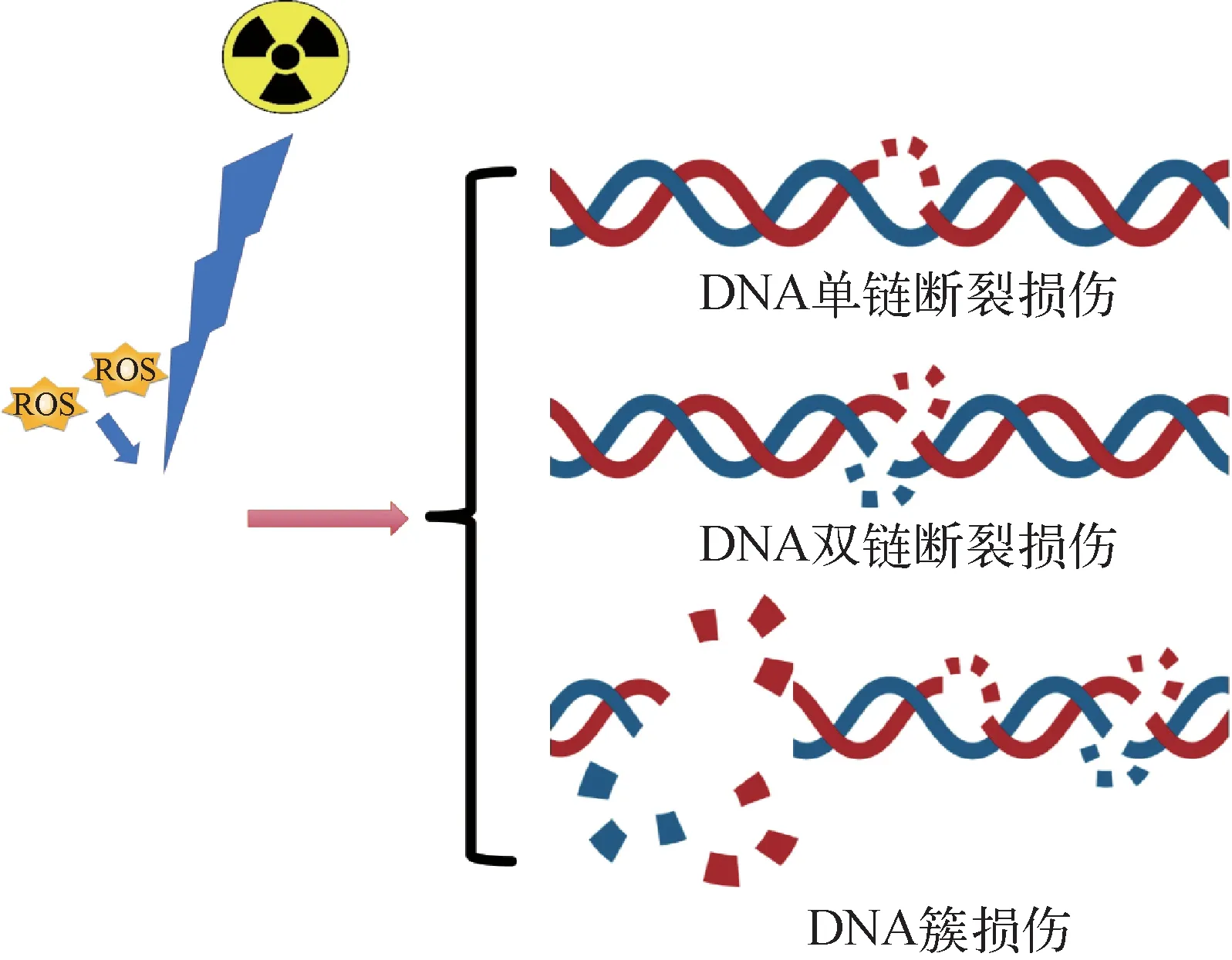
图1 电离辐射诱导的DNA损伤示意图[28-30]Fig.1 Schematic diagram of DNA damage induced by ionizing radiation[28-30]
放疗通常不可避免会让癌细胞对辐射暴露产生抗性,放疗抵抗(radiotherapy resistance, RR)是肿瘤放疗过程中面临的最大挑战,会直接导致抗肿瘤治疗效果明显降低[34-35]。在肿瘤发生放疗抵抗的过程中,肿瘤组织中一定比例的细胞不仅获得更高的放疗抵抗性,而且还会变得更具侵袭性,极容易发生淋巴结和远处转移[36]。放疗抵抗的发生与许多因素有关,包括肿瘤微环境、免疫系统、肠道菌群、营养状况和心理状况等[31,37-39]。然而,DNA损伤和修复还是最主要和内在的因素,是调控癌细胞周期停滞和细胞命运(死亡或存活)最关键的因素[25,31]。由此可见,电离辐射对癌细胞的杀伤能力主要取决于辐射诱导的DNA损伤的程度,肿瘤细胞的DNA损伤反应和肿瘤细胞修复DNA损伤的能力是决定癌细胞结局的关键。
在过去几十年中,通过DNA损伤途径增强肿瘤对电离辐射的反应一直是放射生物学研究中的焦点问题。不同LET的射线沉积于肿瘤细胞DNA分子中能量的差异是导致其不同生物学效应最主要的因素。低LET射线如X射线和γ射线辐照后肿瘤细胞可能只会发生少量的DNA损伤,如碱基和核糖损伤、交联、单链断裂和双链断裂等,这些损伤可能会被短时间内快速修复[40-41]。一些高LET射线如质子、重离子等可能会更多地诱导肿瘤细胞DNA发生大量的成簇损伤,大量的成簇DNA损伤的发生在随后可能将产生更高的染色体重排和致死风险,这也是高LET射线具有更高相对生物效应的主要原因[42-44]。一旦形成复杂的DNA损伤,修复就会进行的非常缓慢,损伤的积累会造成染色体异常,染色体的异常会导致细胞死亡或有丝分裂延迟。
3 电离辐射导致的细胞结局
电离辐射作用于机体后,除了会导致DNA的损伤以外,在分子水平上,辐射产生的自由基也会损害脂质和蛋白质等其他生物大分子,这些分子学上的改变可能会进一步导致细胞器的损伤,这些分子和细胞器的损伤会激活细胞稳态的应激反应,比如DNA损伤反应、未折叠蛋白反应和自噬等[45-46]。当电离辐射诱导的这些损伤有限时,这些过程可能能够确保受辐射细胞的存活,使它们重新进入细胞周期,但是当损伤不能通过修复机制解决时,细胞应激的分子机制从细胞保护模式转变为细胞抑制或细胞毒性模式。因此,不同剂量、不同种类的电离辐射作用于不同细胞可能会造成细胞发生调节性细胞死亡(凋亡、坏死、焦亡、铁死亡等)或细胞应激反应(有丝分裂灾难、自噬、衰老等),图2显示了放射治疗在肿瘤细胞中诱导的不同的细胞命运[26,47-49]。

图2 放射治疗在肿瘤细胞中诱导不同的细胞命运[26,47-49]Fig.2 Radiation therapy induces different cell fates in tumor cells[26,47-49]
3.1 细胞凋亡
细胞凋亡(apoptosis)是由Kerr等[50]在1972年首次提出。凋亡是一种基因调控的、能量依赖的、主动的、程序性的细胞死亡过程,伴随着细胞体积收缩、固缩和凋亡小体的形成,最终以吞噬周围细胞而结束[51]。由于凋亡的存在,质膜的代谢活性和完整性在一定程度上得以保持,而不会引起细胞周围的炎症。凋亡的启动主要由内源性和外源性两种不同的途径激活,两者最终都通过激活执行途径完成凋亡。内源性凋亡是一种由细胞内或细胞外微环境的扰动引发的细胞死亡,以线粒体外膜透化为特征,最终由胱天蛋白酶(Caspases,主要是Caspases-3)执行,外源性凋亡通过质膜受体检测细胞外微环境的扰动而启动,并通过胱天蛋白酶-8(Caspases-8)介导的机制激活最终的执行途径[52]。
在电离辐射后,细胞凋亡的启动涉及这两种激活途径。电离辐射可以激活细胞内促凋亡和抗凋亡的B淋巴细胞瘤-2(B-cell lymphoma-2, Bcl-2)家族成员如BCL2相关凋亡调节因子X(BCL2 associated X, Bax)和BCL2拮抗因子(BCL2 antagonist/killer, Bak)改变线粒体膜的通透性,增加并释放促凋亡因子到细胞质中,从而触发一系列凋亡级联反应[53]。外源性途径由死亡配体-受体特异性结合介导形成构建体,死亡配体-受体结合形成死亡诱导信号复合物(death-inducing signaling complex, DISC),激活Caspases-8并进一步激活Caspases-3、Caspases-6和Caspases-7[54]。尽管不同的细胞类型具有不同的辐射敏感性,但辐射后细胞死亡的模式通常是剂量依赖性,低剂量倾向于诱导细胞凋亡,高剂量更倾向于诱导细胞坏死[26]。
3.2 细胞坏死
细胞坏死(necroptosis)的发生与细胞膜上死亡受体的激活有关,激活的死亡受体可以激活细胞中的受体相互作用蛋白激酶-1(eceptor-interacting protein kinase-1, RIPK1),进一步招募细胞内受体相互作用蛋白激酶-3(receptor-interacting protein kinase-3, RIPK3)形成坏死复合物,随后激活RIPK3,活化的RIPK3可以磷酸化混合谱系激酶结构域样假激酶(mixed lineage kinase domain-like pseudokinase, MLKL),其与紧密连接蛋白共运输至细胞外周,并稳定地结合在质膜上以触发细胞坏死[55]。坏死细胞膜通透性的增加会引起细胞肿胀、细胞器变形,并最终破裂,从而释放促炎物质,坏死细胞中的ATP水平降低,以至于细胞的生理活性无法维持,染色质在坏死过程中随机降解为不同大小的DNA片段[55]。
众多的研究表明,放射治疗可以通过诱导肿瘤细胞坏死来抑制肿瘤的发展。在放疗后死亡的肿瘤细胞中,各种坏死相关蛋白,尤其是MLKL的水平显著上调[56-57]。同样,在放射治疗前用坏死性凋亡抑制剂对肿瘤细胞进行预处理,与未经预处理的直接放射治疗相比,导致明显更少的死亡细胞,这证实了放射治疗可以诱导肿瘤细胞坏死性凋亡[56]。然而,放疗诱导的细胞坏死机制仍有待探讨。近期研究表明,放疗诱导的肿瘤细胞坏死与细胞内左手螺旋DNA(Z-DNA)结合蛋白1(Z-DNA binding protein 1, ZBP1)表达的上调有关[58]。ZBP1是一种核酸传感器,介导宿主对某些病毒的防御,细胞内ZBP1检测到病毒产生的RNA,激活RIPK1和MLKL,引发坏死性凋亡[59]。Yang等[58]的报道表明,放射治疗能够上调肿瘤细胞中ZBP1的表达,激活RIPK1/RIPK3/MLKL信号通路,从而诱导肿瘤细胞凋亡。此外,放疗诱导的肿瘤细胞坏死可能与细胞内线粒体ROS水平上调有关,有研究表明,增加肿瘤细胞中线粒体ROS水平可以通过RIPK1/RIPK3信号诱导坏死性凋亡[60]。然而,有关辐射诱导的坏死和细胞内线粒体活性氧水平增加之间的联系尚不完全清楚,有待进一步的研究探索。
3.3 细胞自噬
自噬(autophagy)是溶酶体介导的细胞降解的一种途径,是一种分解代谢过程,可回收细胞内成分以维持新陈代谢和存活[61]。磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase, PI3K)-蛋白激酶B(protein kinase B, AKT)-哺乳动物雷帕霉素靶蛋白复合物1(mammalian target of rapamycin complex 1, mTORC1)通路在调节自噬中发挥着重要作用[61]。自噬的机制是细胞在应激状态下对有害外界刺激的适应,当细胞面临特定的病理生理或强刺激(如高剂量辐射暴露)时,有利于细胞的存活,然而,过度的自噬会引发细胞死亡。自噬对癌症发展的影响复杂且矛盾,癌症中的自噬已既有可能是肿瘤抑制因子也有可能是肿瘤促进因子[61]。
放疗可诱导肿瘤细胞发生保护性自噬和自噬性细胞死亡,p53、p73等基因在这一过程中起着重要作用[62]。放疗可以通过直接和间接的方式造成DNA损伤,同时会产生大量的ROS,ROS可以诱导细胞发生保护性自噬,活化的保护性自噬可以清除受损的线粒体,降低细胞氧化应激,提高肿瘤细胞对辐射的抵抗力[24]。Mo等[63]的研究发现,Rad51表达降低可抑制鼻咽癌细胞自噬,增加放射敏感性,Wang等[64]发现SMAD4基因突变可通过促进自噬增加胰腺癌的放疗抵抗。另一方面,电离辐射后肿瘤细胞的自我更新能力被破坏,细胞有丝分裂受阻,这可能会刺激细胞发生过度自噬,导致自噬细胞死亡[24]。Djuzenova等[64]发现抑制PI3K-AKT-mTORC1通路并诱导自噬后,胶质母细胞瘤的放射敏感性增加。Palumbo等[65]的研究发现,胶质母细胞瘤细胞系T98G在用电离辐射与替莫唑胺组合治疗后表现出高放射敏感性,这与自噬的激活有关,在抑制自噬后,T98G细胞的放射敏感性下降,表明放疗可以通过自噬性细胞死亡发挥抗癌作用,用雷帕霉素诱导自噬后,T98G的放射敏感性也增强。放疗后肿瘤细胞会发生不同形式的自噬,自噬激活的强度和自噬的靶点是影响放疗后肿瘤细胞命运的关键因素,深入了解不同形式的自噬可能是未来肿瘤个性化治疗的重要依据。
3.4 细胞焦亡
细胞焦亡(pyroposis)是一种严重依赖焦孔素(gasdermin, GSDM)蛋白家族成员的程序性细胞死亡形式[66]。GSDM家族成员具有两个特征保守结构域:N-末端成孔结构域(N-terminal pore-forming domain, N-PFD)和C-末端抑制结构域(C-terminal repression domain, C-RD),在生理条件下,C-RD与N-PFD相互作用并抑制N-PFD活性,当细胞受到刺激时,该结构可以被Caspase依赖性或非依赖性途径切割,并释放细胞毒性N-PFD,N-PFD在膜中寡聚并形成膜孔,会导致炎症介质人白细胞介素1β(Interleukin-1β, IL-1β)和人白细胞介素18(Interleukin-18, IL-18)的释放和焦亡的发生[66]。
放射治疗可以诱导肿瘤细胞发生焦亡。Liu等[67]的研究发现,在10 Gy和20 Gy照射下,骨髓源性巨噬细胞中Caspase-1的活性和焦亡比例增加。Zhang等[68]的研究发现,肺腺癌的放疗可诱导肿瘤细胞焦亡而发挥抗肿瘤作用,放疗破坏了肿瘤细胞DNA,激活了黑素瘤缺乏因子2(absent in melanoma 2, AIM2),并随后诱导了Caspase-1/焦孔素D(gasdermin D, GSDMD)途径介导的焦亡,并明显抑制了肿瘤的生长。GSDMs可被多种细胞凋亡相关的半胱天冬酶切割,并在从细胞凋亡到焦亡的转变中发挥作用[66]。Tan等[69]的研究发现,结直肠癌对放疗的耐药性与GSDME的低表达有关,放疗在GSDME上调后可通过Caspase-3/GSDME途径诱导细胞死亡。这些研究表明,焦亡也是电离辐射诱导细胞死亡的一种重要形式,但是哪种细胞更容易被诱导尚不清楚,需要进一步的研究与探索。
3.5 铁死亡
铁死亡(ferroptosis)是一种新型细胞死亡模式,其发生依赖于铁,表现为脂质过氧化积累并最终导致细胞的死亡,铁死亡的发生主要由谷胱甘肽过氧化物酶4(glutathione peroxidase 4, GPX4)调控,由氧化应激触发,并可被铁螯合剂和亲脂性抗氧化剂所抑制[70]。最近它被认为是肿瘤抑制的重要机制,铁死亡的过程独立于传统的凋亡、坏死等细胞死亡机制,形态学上主要表现为线粒体的改变,包括皱缩、高电子密度的超微结构、嵴减少或消失等[70]。铁死亡的发生与活性氧积累和铁依赖性脂质过氧化密切相关,在细胞内,不稳定的游离铁可以通过芬顿反应产生羟基自由基(hydroxyl radical, ·OH),促进脂质过氧化,铁也作为促进脂质氧化的酶中的辅因子促进铁死亡[70]。
自Lang等[71]首次报道放射治疗可诱导肿瘤细胞铁死亡后,越来越多的研究证实电离辐射诱导的细胞死亡中有着铁死亡的存在[26,72-73]。放疗杀死的肿瘤细胞中观察到多种与铁死亡相关的形态学特征,包括肺、乳腺、食管、卵巢、肾细胞癌、纤维肉瘤和黑色素瘤的细胞[49]。此外,通过放射疗法杀死的肿瘤细胞表现出与铁死亡相关的分子特征,例如肿瘤细胞内脂质过氧化标记物水平,如丙二醛和4-羟基壬烯醛(4-hydroxynonenal, 4-HNE)和铁死亡标记物基因,如前列腺素内过氧化物合酶2(prostaglandin-endoperoxide synthase 2, PTGS2)和酰基辅酶a合成酶长链家族成员4(acyl-CoA synthetase long-chain 4, ACSL4)的上调[72-73]。同样,铁死亡抑制剂或铁螯合剂去铁胺(deferoxamine, DFO)可以减轻辐射诱导的肿瘤细胞的损伤并恢复细胞存活,进一步证实了放疗诱导的肿瘤细胞死亡中有铁死亡的发生[72]。也有研究表明,电离辐射能够通过抑制溶质载体家族7成员11(solute carrier family 7 member 11, SLC7A11)对胱氨酸的摄取来促进铁死亡,导致癌细胞中脂质过氧化的发生,而这种脂质过氧化可被干扰素γ((interferon γ, IFN-γ)进一步加剧[71]。放疗过程中ROS的增加可诱导激活核因子-红细胞2相关因子2(nuclear factor erythroid-2 related factor 2, Nrf2)-血红素加氧酶-1(heme oxygenase-1, HO-1)途径[74]。Nrf2-HO-1通路在铁死亡中可能发挥着相反的作用。Feng等[75]的研究发现Nrf2可激活SLC7A11抑制缺铁性贫血,并降低食管鳞状细胞癌的放射敏感性,有研究却发现,激活的Nrf2-HO-1通路可以提高Fe2+水平并诱导结直肠癌癌症细胞的铁死亡[76]。可见,铁死亡也是辐射诱导细胞死亡的一种重要方式。
3.6 有丝分裂灾难
有丝分裂灾难(mitotic catastrophe, MC)通常是指由于物理或化学因素导致过早或不适当进入分裂阶段的一系列事件,这些因素在辐射诱导的细胞死亡中起着特定作用[77]。导致这种有丝分裂灾难的因素包括广泛的DNA损伤、有丝分裂机制的功能障碍、微管的异常稳定性和周期检查点缺陷等。诱导致命有丝分裂灾难的主要途径有两种:第一种是p53基因的下调导致G2或M期检查点的抑制,使DNA损伤无法修复;第二种是中心体超扩增,过度膨胀的中心体会导致多极分裂、异常染色体分离和细胞核异常(巨核、双核或多核等)[77]。当有丝分裂灾难发生时,细胞在短暂的G2停滞后继续有丝分裂,然后多个有丝分裂检查点蛋白的表达增加,并促进有丝分裂延迟或停滞,在此期间,延迟凋亡可能在中期被激活[77]。有研究表明,Caspase-2在有丝分裂灾难后的延迟凋亡中起着关键作用[78]。然而,一般来说,细胞不会在分裂中期死亡,因为它们可以适应有丝分裂检查点,细胞继续经历一个或多个细胞周期,并获得越来越多的染色体畸变,成为非整倍体或多倍体,最终,这些细胞由于延迟凋亡、坏死或进入细胞衰老而死亡[79]。
在放射治疗后,可以观察到由于有丝分裂灾难引起的细胞死亡[26,47]。肿瘤细胞在放疗诱导的DNA损伤后很容易经历有丝分裂灾难,Fragkos等[80]发现p53基因失活后,放疗既不激活p21基因也不抑制细胞周期蛋白依赖性激酶1(cyclin-dependent kinase 1, CDK 1)-细胞周期蛋白A(cyclin A)复合物,导致G2或M检查点失活和过早进入有丝分裂。p53基因对DNA修复也很重要,过早进入有丝分裂的细胞将携带未修复的DNA损伤,促进有丝分裂灾难的发生[47]。有报道表明,放疗诱导的有丝分裂灾难后的延迟死亡通常发生在放疗后2~6 d[81]。Cheng等[82]的研究发现B12536联合2 Gy放疗在体外和体内均可诱导口腔癌细胞有丝分裂灾难性死亡而增加放疗敏感性。而Gordon等[83]也发现LB100联合放疗能够通过抑制蛋白磷酸酶2A(protein phosphatase 2A, PP2A)增强胶质母细胞瘤中辐射诱导的有丝分裂灾难而增加其放射敏感性。
3.7 细胞衰老
Hayflick和Moorhead在20世纪60年代早期首次报道了细胞衰老(senescence),细胞衰老表现为永久的增殖停滞,并伴随着多种表型的变化[84]。细胞衰老涉及细胞周期停滞和具有自分泌、旁分泌和内分泌活性的炎性细胞因子释放,衰老细胞也表现出形态学改变,包括扁平的细胞体,细胞质中的空泡化和粒度以及异常细胞器,已经鉴定出几种细胞衰老的生物标志物,包括衰老相关β半乳糖苷酶(senescence-associated β-galactosidase, SA-β-gal)、p16基因和p21基因等,但这些标志物都缺乏敏感性和组织特异性[85]。衰老相关表型的激活会进一步放大细胞增殖停滞的影响,并导致受损的组织再生、慢性年龄相关疾病(如癌症、动脉粥样硬化、骨关节炎和神经退行性疾病)和机体衰老[86-87]。细胞衰老可能发生在受调控的不同生物过程中,如胚胎发育,也可由细胞损伤诱导,如DNA损伤、端粒缩短或功能障碍、癌基因激活或肿瘤抑制功能丧失、表观遗传变化和细胞器损伤等[88]。衰老应激的主要原因是DNA损伤,它激活DDR和典型的p53-p21通路[89]。
细胞衰老也是放疗过程中电离辐射诱导的一种常见的非致死性细胞结局。有研究表明,电离辐射损伤肿瘤细胞DNA后,活化的p53基因上调p21基因的表达,进而抑制CDK-cyclin复合物的表达,抑制CDK 1-cyclin A复合物会将细胞阻滞在G2或M期,抑制CDK 2-cyclin E/A复合物和CDK 4-cyclin D复合物可促进磷酸化视网膜母细胞瘤(phosphorylation retinoblastoma, pRB)的去磷酸化,并将细胞阻滞在G1或S期[90-91]。此外,电离辐射诱导的细胞内ROS和蛋白激酶C(protein kinase C, PKC)的升高也能增加p16的表达,抑制CDK-cyclin复合物并促进pRB去磷酸化,导致肿瘤细胞衰老[92]。细胞衰老在癌症放疗中起着双重作用。一方面,由电离辐射诱导的细胞衰老可以抑制肿瘤细胞的增殖并激活癌症免疫监视[93-94]。另一方面,尤其是在放疗的后期,衰老细胞的积聚可能对预后产生不利影响,因为衰老细胞分泌的细胞因子所形成的免疫微环境可能促进肿瘤复发,一些癌症在放疗后的高复发率可能与衰老细胞的积累有关[90]。
4 总结与展望
核技术在医学领域有着广泛的应用,明确电离辐射所产生的生物学效应对安全高效利用核技术至关重要。电离辐射的直接或间接作用导致的DNA等分子的损失可能会进一步诱导细胞发生凋亡、坏死、焦亡、铁死亡、有丝分裂灾难、自噬、衰老等不同的生物学效应。在电离辐射的放疗应用中,关于放射生物学效应的研究仍有很多问题值得探讨,例如:(1) 随着核技术的不断发展与进步,新型医疗设备和放射性药物作用于机体是否存在新的生物效应,不同类型的放射性核素诱导的细胞死亡方式是否相同;(2) 关于放射性药物内照射相关的生物学研究较少,在临床放射性药物的使用过程中,核素的化学毒性是否可以忽略,化学毒性与辐射毒性的叠加是否会产生新型的生物学效应亟待研究;(3) 电离辐射诱导的细胞死亡是否存在其他死亡方式如铜死亡、巨泡死亡、碱死亡、氧化死亡、网状细胞死亡等尚待研究,此外,单纯去干预某一种死亡方式是否就能改变其辐射敏感性,或者干预其中一种死亡方式会不会影响其他类型的死亡,不同死亡方式之间是否存在着转换的关系,这都需要进一步的研究和总结。
