醛糖还原酶和晚期糖化终产物受体对糖尿病视网膜病变神经细胞凋亡、自噬和高尔基应激水平的影响
2023-06-15王雅清李红敏张茜玉王莉王勇刘永胜庞英杰
王雅清 李红敏 张茜玉 王莉 王勇 刘永胜 庞英杰
糖尿病视网膜病变(diabetic retinopathy,DR)是糖尿病最常见和最严重的并发症之一,是老年人视力受损的主要原因[1]。有证据表明,抑制神经细胞凋亡可能是预防DR病理变化的一种治疗策略[2]。体内细胞存活平衡取决于细胞蛋白质和细胞器的合成和降解。如果自噬失败,这可能导致有害的受损细胞器和蛋白质聚集体的积累[3]。自噬受一系列压力和刺激的调节,如内质网和高尔基应激、缺氧、细胞毒性和感染。根据刺激,细胞通过特定元素的自噬降解或与其他途径的相互作用来实现多种功能,从而导致细胞凋亡[4]。因此,细胞死亡和存活取决于自噬控制。自噬途径在防止衰老和某些癌症、感染、神经退行性疾病以及代谢、炎症和肌肉疾病方面起着关键作用[5]。高血糖是神经退行性疾病发展的关键因素,醛糖还原酶(aldose reductase,AR)表达水平升高与DR发病机制有关[6]。此外,由于AR在高血糖患者葡萄糖代谢中的活性增强,活性氧和山梨糖醇的产生增加,表明AR在神经退行性疾病的进展中发挥了作用[7]。晚期糖化终产物受体(advanced glycation end product receptor,RAGE)免疫球蛋白家族跨膜蛋白是细胞外糖基化终产物AGE 以及其他类别的蛋白质和分子的主要受体[8]。RAGE的最佳特征配体是CML蛋白加合物,其在高血糖和其他压力条件下升高。RAGE是第一个发现的也是迄今为止研究最多的 AGE受体[9]。临床证据表明,高RAGE水平可能会促使视网膜毛细血管的增殖变化,使其发生更严重的DR[10]。本研究在1型链脲佐菌素诱导的糖尿病小鼠模型中探讨AR和RAGE对DR神经细胞凋亡、自噬和高尔基应激水平的影响。
1 材料与方法
1.1 实验材料
1.1.1 实验小鼠:使用C57BL/6J 小鼠(Jackson Laboratories,Sacramento,CA,USA)购自河北省实验动物中心,合格证号:SCXK(冀)2022-001。将其饲养在 12 h光照/黑暗循环中,光照水平<60勒克斯。饲养期间小鼠可随意获取食物和水。动物研究符合 ARVO 关于在眼科和视觉研究中使用动物的声明。该研究方案得到了当地动物研究伦理委员会的批准。
1.1.2 糖尿病小鼠模型:通过连续5 d腹膜内注射链脲佐菌素(55 mg/kg BW),在2个月大的禁食雄性小鼠中诱发胰岛素缺乏型糖尿病。根据需要给予胰岛素以维持 BW并允许 BW 缓慢增加,同时允许高血糖、多尿和食欲亢进(每2~3天0~0.2 U)。每 2~3个月通过测量总糖化血红蛋白水平和测量血糖浓度。血糖值超过 300 mg/dl 的小鼠被认为患有糖尿病。
1.1.3 实验分组:对照组(标准条件饲养的小鼠,注射0.9%的氯化钠溶液作为对照,n=15),模型组(如前所述,通过腹腔注射链脲佐菌素诱导小鼠发生糖尿病,n=15),观察组(糖尿病小鼠诱导成功后,饮食中加入依帕司他原药按50 mg/kg,FPS-ZM 5 mg/d喂养,依帕司他和FPS-ZM分别为AR和RAGE的抑制剂,用药持续1周,n=15)。
1.2 方法
1.2.1 蛋白质印迹分析:从安乐死的小鼠中快速解剖视网膜组织并在含有蛋白酶抑制剂(Beyotime)和磷酸酶抑制剂的 RIPA 缓冲液(Beyotime,上海,中国)中裂解。裂解液于 4℃ 12 000 r/min 离心 10 min,收集上清液。通过十二烷基硫酸钠聚丙烯酰胺凝胶电泳(SDS-PAGE)分离等量的蛋白质(20 μg)并印迹到聚偏二氟乙烯膜(Millipore,USA)上。5%脱脂奶粉室温封闭2 h后,膜与以下一抗4℃孵育过夜:RAGE、AR。在 Tris 缓冲盐水和 Tween-20(TBST)中洗涤后,将免疫印迹与辣根过氧化物酶偶联的二抗(1∶10 000,Cell Signaling Technology)在室温下孵育 1 h,使用化学发光试剂使用化学发光试剂显色印迹。增强化学发光(ECL)试剂盒(Millipore)。
1.2.2 RT-PCR:为了分析 mRNA 表达,从小鼠视网膜组织中提取总RNA,定量并使用 1μg 总 RNA 合成第一链 cDNA。使用 StepOnePlusTM实时 PCR 系统进行实时(RT)-PCR。PCR 扩增使用 SYBR Green PCR Master Mix 进行三次重复。使用以下循环优化每个基因的反应条件:初始循环设置为 95°C 7min;在 95°C15秒和 60°C 1min下进行 30 个循环;在 95°C1min、55°C30秒和 95°C30秒下进行1个循环以获得解离曲线。不含模板 cDNA 的反应混合物用作阴性对照。使用GAPDH的 Ct 值作为内源对照,通过比较 Ct 方法(ΔΔCT)确定基因表达水平。
1.2.3 视网膜电图(ERG):在患糖尿病后,对各实验组小鼠进行暗视和明视 ERG 测试。对于暗视 ERG 记录,向小鼠呈现强度增加的单次闪光(2.19 × 10-4至 83.7 cd·s-1·m-2,0.6 log 单位步长),每次重复5次,刺激间隔为 20 s最亮的闪光为 1 min。平均每个闪光亮度下的5个 ERG 轨迹。从背景开始≥15 min后,通过将测试闪光(0.016~377 cd·s-1·m-2)叠加在杆饱和强度(30 cd/m2)的稳定背景上,获得孤立的锥体成分。
1.2.4 视网膜组织中的免疫荧光:将眼球摘除并用 4% 多聚甲醛固定,在磷酸盐缓冲液中的 30% 蔗糖中冷冻保护,在包埋培养基中冷冻。10 μm 视网膜切片用 BSA 封闭,并与抗Caspase-8和抗Caspase-3 一起孵育,在 4℃下过夜,与适当的二抗一起孵育。通过省略一抗建立阴性对照。将切片与适当的二抗一起孵育。将切片冲洗并用含有用于细胞核染色的 4’,6’-二氨基-2-苯基吲哚的抗褪色培养基盖玻片。在荧光显微镜下检查载玻片,使用合适的荧光素异硫氰酸酯 1 和罗丹明发射滤光片。来自每只动物的3个非连续视网膜切片(相距 100 μm)和每组4只动物的3张图像被纳入分析。
1.2.5 TUNEL 检测:通过TUNEL测定检查细胞凋亡计数视网膜的 TUNEL 阳性细胞核 GCL。简而言之,在室温下用冰-丙酮溶液固定 8 min后,将冷冻保存的组织切片与 1 ml 封闭缓冲液(PBS 中的 3% 正常山羊血清)一起孵育 1 h。孵育后,将切片置于透化溶液(0.1% Triton X-100 的 0.1% 柠檬酸钠溶液)中冰上 2 min。将切片在 50 μl TUNEL 反应混合物(Roche Diagnostics GmbH,德国)中孵育,并在 37℃下在黑暗中孵育 60 min。在反应后用 4,6-二脒基-2-苯基吲哚(DAPI)对切片进行复染。在荧光显微镜(Olympus,Japan)下观察切片。
1.2.6 酶联免疫吸附试验:使用相应的小鼠 TNF-α、IL-6、IL-1β DuoSet ELISA 开发试剂盒(R&D Systems,Inc.,Minneapolis,MN,USA)和小鼠 VEGF DuoSet 试剂盒(R&D Systems,Inc.)测定培养基中分泌的 TNF-α TNF-α、IL-6、IL-1β和 VEGF。使用 BioTek SynergyTM4 Hybrid Microplate Reader 检测光密度,并通过平行生成的标准曲线外推的吸光度值推导出细胞因子水平。
2 结果
2.1 蛋白印迹分析AR和RAGE表达 模型组较对照组AR和RAGE的蛋白表达升高(P<0.05),观察组较模型组AR和RAGE的蛋白表达降低(P<0.05),表明AR和RAGE促进了DR的发生,而药物阻断成功抑制体内AR和RAGE的表达水平。见图1,表1。
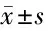
表1 蛋白印迹分析AR和RAGE表达

图1 蛋白印迹分析AR和RAGE
2.2 AR和RAGE诱导自噬机制的启动 通过PCR分析大鼠视网膜裂解组织中自噬相关蛋白Beclin-1、LC3 II/I 和 p62/SQTSM1的mRNA表达,模型组较对照组Beclin-1、LC3 II/I 和 p62/SQTSM1的mRNA表达升高(P<0.05),观察组较模型组表达降低(P<0.05),实验数据表明AR和RAGE参与了自噬通量功能障碍的形成。见表2。
表2 PCR分析自噬蛋白的转录
2.3 AR和RAGE参与DR的发生 模型组较对照组暗视b波和暗视a波的振幅降低(P<0.05),观察组较模型组升高(P<0.05),实验数据表明当AR和RAGE表达受阻降低了链脲佐菌素诱导的小鼠DR。见表3。
表3 ERG分析小鼠的视网膜受损程度 μV,
2.4 AR和RAGE加剧神经细胞凋亡 通过免疫荧光分析大鼠视网膜组织中Caspase-8和Caspase-3的蛋白表达,模型组较对照组Caspase-8和Caspase-3的蛋白表达升高(P<0.05),观察组较模型组的蛋白表达降低(P<0.05),表明当降低AR和RAGE的表达,caspase 8、Caspase-3的活性和细胞凋亡就会得到改善,从而保护细胞免于死亡。TUNEL分析了小鼠视网膜神经细胞的凋亡,与对照组相比,链脲佐菌素诱导的DR神经细胞凋亡量增加(P<0.05),当AR和RAGE受到抑制后,阳性细胞数量降低(P<0.05)。见图2,表4。
表4 免疫荧光分析Caspase-8和Caspase-3表达

观察组 模型组 观察组
2.5 ELISA试剂盒测定分泌的炎性因子和VEGF水平 通过ELISA分析血清炎性因子TNF-α、IL-6、IL-1β及VEGF的分泌水平,模型组较对照组TNF-α、IL-6、IL-1β及VEGF的水平升高(P<0.05),观察组较模型组降低(P<0.05)。见表5。
表5 ELISA分析TNF-α、IL-6、IL-1β及VEGF水平 pg/ml,
2.6 PCR分析高尔基应激反应相关因子的表达 模型组较对照组DR4、TRAPPC13、MAPK和ETS的mRNA表达升高(P<0.05),观察组较模型组表达降低,差异有统计学意义(P<0.05),数据表明抑制AR和RAGE表达可以降低小鼠DR神经细胞的高尔基应激反应。见表6。
表6 PCR分析高尔基应激反应相关因子的表达 pg/ml,
3 讨论
DR被认为是一种微血管并发症。最近的研究指出,神经变性发生在血管病变发生之前,并有助于DR的发生和进展[11]。AR是多元醇途径的第一个限速酶和醛酮还原酶超家族的成员,负责 NADPH 依赖性葡萄糖还原为山梨糖醇[12]。一般来说,葡萄糖主要通过糖酵解途径代谢,因此多元醇途径对葡萄糖代谢的影响很小,不被认为对非糖尿病患者构成威胁。然而,在高血糖时,多元醇途径和AR刺激后有很大的增加[13]。依赖RAGE和AR的信号传导是高血糖积累的致病原因,并且其本身对于正常发育和衰老在很大程度上是可有可无的。因此,RAGE 途径的抑制构成了治疗与AGE积累相关的疾病的有吸引力的目标。在生理上,几种RAGE抑制剂是通过选择性剪接产生的,以产生没有细胞内结构域的RAGE分子[14]。已经产生了几种RAGE抑制剂药物,其中一些已进入临床研究,包括 PF-04494700,目前正处于治疗阿尔茨海默病患者痴呆症的3期试验中。PF-04494700 通过阻止许多配体与 RAGE结合而在细胞外发挥作用,并且能够穿过血脑屏障[15]。另一种细胞外作用的RAGE抑制剂 FPS-ZM1 能够阻断由 Aβ42 和 BSA-AGE 介导的RAGE信号传导。其他靶向RAGE细胞内结构域的药物正在开发过程中,该结构域与RAGE依赖性信号调节剂 DIAPH1 相互作用,尽管这些研究仍处于临床前阶段。
自噬是在正常和压力条件下调节细胞稳态的重要过程。多项研究表明,自噬的作用会随着时间和损伤程度而变化,自噬早期可能通过抑制细胞凋亡而具有保护作用,而晚期或过度自噬会加重细胞损伤[16]。一项研究发现适当的自噬的诱导可能对果蝇和帕金森病小鼠产生神经保护作用[17]。我们的初步研究表明,暴露于高糖条件下的视网膜神经细胞的自噬水平升高,自噬的激活可能表现出细胞保护作用。多项研究表明,自噬功能障碍是DR发病机制中存在的早期事件[18]。在本研究中,我们发现在糖尿病小鼠模型中,自噬相关蛋白 LC3-II 和 Beclin-1 的表达升高,而降低RAGE和AR的表达处理有效地抑制自噬和自噬相关蛋白的表达。尽管自噬在DR发病机制中的作用仍不确定,但现有的几项研究表明,抑制高糖诱导的细胞和神经元的自噬,同时激活自噬可产生保护作用。基于此,我们推测 DR中视网膜自噬的恢复可能会发挥保护作用。
这项研究检查了由于链脲佐菌素诱导的糖尿病在小鼠视网膜中发生的电生理、和生化变化。除了已知的细胞凋亡过程外,自噬途径可能与神经元视网膜变性有关。总体而言,我们的结果表明,视网膜神经特别容易受到糖尿病的影响,这可能是由于它们的高代谢需求。我们的结果显示暗视a波和b波减少,这与其他作者先前报道的糖尿病小鼠模型数据一致。在体内研究中,糖尿病小鼠表现出Beclin-1和p62/SQTSM1的明显增加,并伴有视网膜组织中溶酶体功能的损害。这表明溶酶体损伤和自噬功能障碍是DR发病机制中存在的早期事件。正如其他小组先前所描述的那样,视网膜缪勒氏细胞会对各种损伤做出反应,包括向视网膜组织大量释放 VEGF。这种后来的现象可能导致血视网膜屏障的破坏,最初导致视网膜水肿,导致视网膜新生血管形成[19]。蛋白质 p62/SQTSM1 是一种功能性蛋白质,它在细胞的命运中起到关键作用。在本研究中,我们发现Caspase-8和Caspase-3参与高血糖条件下诱导的视网膜神经细胞的凋亡。p62/SQTSM1 和Caspase-8和Caspase-3途径之间的相互作用表明 p62/SQTSM1 决定了糖尿病条件下的神经细胞的死亡。我们的研究结果表明,在高糖条件下,自噬机制被触发,但自噬通量受到损害。由于其溶酶体储存性质,自噬底物(p62/SQTSM1)在细胞质中积累,发出细胞凋亡和大量 VEGF 产生的信号。当RAGE和AR降低表达恢复溶酶体活性,改善自噬并减轻视网膜细胞凋亡和VEGF产生。VEGF诱导的新血管形成是DR的另一个主要原因,其涉及不稳定的视网膜毛细血管的异常增殖。血浆内容物毛细血管渗漏到视网膜会导致视网膜水肿和视力障碍。研究表明,RAGE和AR抑制是减少DR中视网膜炎症和 VEGF 分泌的有吸引力的治疗靶点。TNF-α是DR中最主要的促炎细胞因子之一,参与白细胞活化和细胞凋亡,主要由活化的内皮细胞和炎症细胞产生。与我们的研究结果表明,先前的研究表明,糖尿病视网膜中TNF-α、IL-6、IL-1β 的表达增加,并有助于 DR 的进展。表明RAGE和AR抑制能抑制炎症介质的释放和减少细胞凋亡因子的表达来改善视网膜损伤。
类似于内质网应激,高尔基体应激也可以诱导细胞死亡。ARF4 是一种小 G 蛋白,调节来自高尔基体的囊泡运输。使用布雷菲尔德菌素A处理后,ARF4 mRNA 水平增加,表明 ARF4 是CREB3 途径的靶基因。CREB3 通路的其他靶基因包括DR4和TRAPPC13[20]。DR4 编码一种死亡受体,调节高尔基体应激诱导的细胞死亡,而TRAPPC13是TRAPPIII 复合物的组分,在某些应激条件下调节自噬通量。他们将 CREB3 鉴定为定位于内质网膜的传感器转录因子,它属于 ATF6 蛋白家族。ATF6 是众所周知的传感器转录因子,调节内质网应激反应的 ATF6 通路,其中靶基因包括内质网伴侣。在没有高尔基体应激的情况下,CREB3 保留在ER膜中,而CREB3 在高尔基体应激时被转运到高尔基体并被蛋白酶S1P和S2P裂解,导致 CREB3 的胞质区域从高尔基体膜及其易位到细胞核。除了 CREB3 通路之外, MAPK-ETS 通路也诱导细胞死亡以应对高尔基体应激,通过检查在高尔基体胁迫诱导剂处理后转录增加的靶基因的启动子区域,并通过转录因子结合基序富集分析寻找可以与启动子结合的转录因子,从而得到 鉴定来自 ETS 家族的三种转录因子(ELK1、ETS1 和 GABPA/B)。该项研究还发现MEK1/2 和ERK1/2 等MAPK被 BFA、Golgicide A 和莫能菌素处理激活,并提出高尔基体应激通过激活 MAPK 级联反应激活 ETS转录因子,导致细胞死亡。[20]
综上所述,我们研究表明,AR表达水平与DR发病机制相关,RAGE的最佳特征配体是CML蛋白加合物,其在高血糖和其他压力条件下升高。1型链脲佐菌素诱导的糖尿病小鼠模型中AR和RAGE对DR神经细胞凋亡、自噬和高尔基应激水平有影响。抑制RAGE和AR可预防链脲佐菌素诱导的小鼠的DR,降低RAGE和AR体内的表达可显著改善小鼠的视力情况,并且可以抑制视网膜病变神经细胞凋亡、自噬和高尔基应激水平的发生。
