抗谷氨酸脱羧酶65抗体阳性相关中枢神经系统病变临床特点分析
2022-09-01侯彦波刘晶瑶
高 颖, 黄 瑶, 侯彦波, 刘晶瑶
1988年《新英格兰医学杂志》发表的一项研究中首次描述了谷氨酸脱羧酶(GAD)抗体[1],GAD抗体是合成抑制性神经递质γ-氨基丁酸的限速酶,与多种神经系统综合征相关,包括僵硬人综合征、边缘性脑炎、癫痫和小脑共济失调等。目前关于GAD抗体相关的综合征并不常见,本文现报道1例抗GAD抗体相关小脑性共济失调(cerebellar ataxia,CA)和1例抗GAD抗体相关的自身免疫性脑炎(autoimmune encephalitis,AE),以加深临床对本病的认识。
1 病例资料
患者1,女,61岁,无业。主诉视物双影1 y,走路不稳9 m。该患者于入院前1 y无明显诱因出现视物双影,左视时明显,自行口服中药治疗后上述症状稍有所好转。9 m前出现走路不稳,表现为行走时不能走直线,步距增宽,左右摇摆,并伴有头晕,病程中无肢体无力,无头痛及恶心、呕吐,无意识障碍、抽搐等。既往:糖尿病病史7 y。入院查体:卧位血压110/70 mmHg,站立位血压108/70 mmHg,神清语明。左侧眼睑下垂,双侧瞳孔直径3.0 mm,对光反射灵敏,双眼活动灵活,双眼可见双向水平眼震。双侧鼻唇沟对称等深,伸舌居中。四肢肌力5级,四肢肌张力正常,双侧腱反射对称引出。感觉检查未见异常。左手指鼻及左侧跟膝胫试验均欠稳准。右侧霍夫曼征阳性,右侧Chaddock征阳性。昂白氏试验阳性。无项强,克氏征阴性。辅助检查:糖化血红蛋白6.6%,抗核抗体系列、女性肿瘤标志物筛查、甲功五项、血常规、生化、梅毒抗体及抗HIV抗体、风湿检测、血沉、布氏杆菌、纤溶测定等均未见明显异常。腰椎穿刺提示脑脊液无色透明,颅压150 mmH2O,蛋白定量 0.42 g/L,脑脊液葡萄糖及氯化物均正常;脑脊液及血清GAD65强阳性,血清抗Sulfatide抗体及抗 GM1抗体弱阳性。影像学检查:头部MRI平扫+弥散及增强扫描:双侧多发腔隙性脑梗死、缺血灶;颈椎MRI、胸椎MRI及腰椎MRI平扫提示脊椎退行性变。肺部CT平扫:双肺下叶限局性肺气肿,右肺上叶结核伴钙化灶。前庭功能:自发眼震描记(+)垂直向下,较强烈,扫视试验正常,凝视试验(+)垂直向下,视跟踪试验Ⅱ-Ⅲ型,视动性眼震试验对称,位置试验未见异常,视动中枢检查未见异常。乳腺及妇科超声、耳镜、腹部超声、骨盆正位片、心电图未见异常。临床诊断:小脑性共济失调(抗GAD65抗体相关),治疗上给予甲强龙冲击治疗(起始量500 mg/d,每3 d减量一半),并序贯减为按体重 1 mg /kg 强的松口服治疗3 w出院。出院后3 m随访患者症状有改善,眼睑下垂好转,视物不清较前缓解,仍走路不稳。
患者2,男,32岁,公司职员。主诉四肢无力5 d,加重伴言语笨拙4 d。该患者于入院前5 d出现四肢无力伴发热,可自行行走,4 d前出现言语笨拙,吐字不清,症状逐渐加重。病程中无抽搐及排尿困难。既往:病前有感染史。查体:体温37.1 ℃,血压140/84 mmHg,神清,构音障碍。双眼球活动自如,双瞳孔等大同圆,直径约4.0 mm,对光反射灵敏。双侧鼻唇沟等深,伸舌居中。四肢肌张力正常,左下肢肌力3级,余肢体肌力4级。四肢腱反射减弱。感觉检查未见异常。双侧病理反射未引出。项强3横指,克氏征阳性。患者入院后仍间断发热,体温最高达40.0 ℃,出现发作性抽搐1次,表现为双眼向左凝视,四肢伸直,无意识丧失、舌咬伤及尿失禁,症状持续约1 min左右缓解。辅助检查:血常规:白细胞7.09X109/L,中性粒比例86.6%,血肌酐:148 μmol/L,BUN 10.7 mmol/L,尿常规:蛋白1+,肝功、血糖、抗核抗体系列、纤溶测定、男性肿瘤标志物、甲功五项、凝血常规、梅毒抗体及抗HIV抗体、风湿检测、血沉、布氏杆菌等均未见明显异常。腰椎穿刺示脑脊液无色透明,颅压180 mmH2O,潘迪氏试验+,白细胞64*106/L,蛋白定量 0.68 g/L,葡萄糖及氯化物正常,脑脊液及血清GAD65抗体阳性,细胞学检查、结核抗体、隐球菌未见明显异常。影像学检查:头MRI平扫+弥散:胼胝体压部异常信号(见图1)。肺CT提示双肺炎症。24 h脑电图:弥漫性慢波活动夹杂低幅混合频率,双侧额颞区稍多量尖化θ波(见图2)。临床诊断:抗GAD65抗体相关性脑炎,治疗上先后给予丙种球蛋白0.4 g/kg每天静脉注射(疗程5 d),甲强龙冲击治疗(起始量500 mg/d,每3 d减量一半并序贯减为口服),以及抗病毒等对症治疗3 w后,患者症状明显减轻,吐字较入院时清晰,可在他人挽扶下站立床旁尚不能行走,无发热及抽搐发作。出院3 m后随访,患者一般状态较好,可正常交流,并独立下床适当活动。
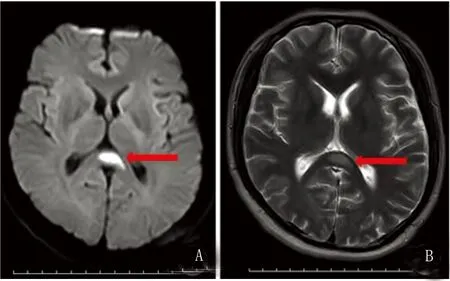
图1 患者2头部MRI+DWI胼胝体压部可见回旋镖状异常信号,DWI(A)、T2WI(B)均呈高信号(红色箭头标注),T1WI呈低信号
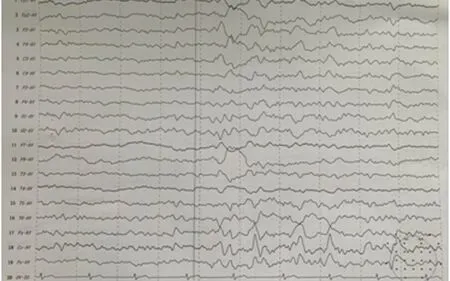
图2 患者2的脑电图可见弥漫性慢波活动夹杂低幅混合频率以及尖化θ波
2 讨 论
GAD抗体以GAD65和GAD67两种构型表达,这两个构型是由不同染色体上两个不同的基因编码,在辅因子结合区域和C末端的结构域中具有71%同源性[2],GAD67主要存在于抑制性神经元的胞浆中,调节GABA的水平;而GAD65锚定在突触囊泡上,能够响应急性需求而瞬时介导GABA合成,并促进含有GABA的突触囊泡从高尔基体转运至突触末端[3]。与GAD67相比,GAD65更常见[4]。2015年Manto等根据一系列相关的临床及动物实验,探讨了GAD65在神经系统中的致病作用,关于抗GAD65抗体的发病机制有如下几个可能:(1)GAD65抗体通过抑制GAD酶活性或阻断GAD65介导的囊泡运输,干扰了小脑中GABA的释放,从而导致了疾病的发生[5];(2)GABA的持续减少会引起谷氨酸释放的增加,从而导致蒲肯野细胞具有明显的过度兴奋性,出现谷氨酸兴奋性中毒;3、不排除T细胞介导的机制参与[6]。抗GAD65抗体是中枢神经系统和非神经系统疾病自身免疫的生物学标志物,GAD65在非神经自身免疫疾病中较为常见,包括1型糖尿病、自身免疫性甲状腺疾病和恶性贫血等,上述疾病的一种或多种可同时存在于约70%的GAD65神经系统自身免疫性患者中[7]。而在神经系统中则相对罕见,包括僵硬人综合征(SPS)、边缘性脑炎、癫痫和小脑共济失调等,可以单独或叠加出现,有文献称之为 “抗GAD抗体综合征”。根据既往文献报道,GAD65抗体累及神经系统不同部位时,其临床表现也各异,本文介绍的CA多见于女性,平均发病年龄在58岁,亚急性起病,逐渐进展,最常见的临床表现是步态共济失调,其次是肢体共济失调、构音障碍和眼球震颤,部分患者还可在发病后数年出现SPS症状。一般在发病前会出现持续性的眩晕,有的也会出现复视或构音障碍[8]。本文中的患者1临床表现与文献报道均相符合。另外,CA的脑脊液细胞数和蛋白多正常,抗GAD65抗体鞘内合成和CSF 特异性寡克隆区带阳性常见,头MRI可显示正常或小脑萎缩[9]。而AE多以急性或亚急性起病,除出现头痛、发热等前驱症状外,还包括癫痫发作,认知和近记忆缺损以及精神行为异常,言语障碍、运动障碍、不自主运动、意识水平下降等表现。行头颅MRI检查通常在T2加权像或FLAIR序列上显示边缘系统或其他区域高信号。脑电图检测发现单侧或双侧额颞区弥漫性慢波及尖慢波对诊断具有一定提示意义[10]。诊断主要依赖于血清及脑脊液抗体的检测。本文报道2例患者临床表现与上述文献相似,且脑脊液及血清中均检测到GAD65抗体,结合相关辅助检查,参照2019年的《免疫介导小脑共济失调的分类及诊断》和2017年的《中国自身免疫性脑炎专家共识》,本文两例患者诊断明确。此外,尽管 GAD65 抗体的自身免疫性通常为非副肿瘤性,但对于肿瘤的可能性也不能完全排除,需积极筛查,虽然这种情况并不常见,但仍要注意此种可能性的发生,本文中患者肿瘤标志物均无明显异常,经过初步筛查未发现肿瘤,仍建议患者进一步行PET-CT等相关检查并定期复诊。GAD65抗体相关疾病除了可与肿瘤合并外,还可与多种自身抗体共存,如抗 NMDAR、抗LGI1、抗 GABAA、GABAB等[11],另外若合并的抗神经元抗体性质为副肿瘤性的,则会提示肿瘤的类型以及可能会与预后相关,而多重抗神经元抗体阳性还会导致临床症状变化或叠加[12]。本报道中的患者1除了GAD65强阳性外,还出现了IgG 型抗体Sulfatide、IgM 型抗体 GM1弱阳性。抗GM1抗体是常见的神经节苷脂抗体,其阳性通常提示运动神经损害多见,而抗Sulfatide 抗体是则是不同于神经节苷脂的一种酸性糖脂,以感觉神经受累多见[13],两者均属于抗周围神经膜表面糖脂抗体,与自身免疫介导的急性和慢性多发性周围神经病有密切关系。那又如何在并发多个自身抗体患者身上排除其他抗体影响,从而建立起GAD65抗体与本病的致病性联系,2020年Graus等就此问题提出了用于临床实践的诊断算法:首先是血清中存在高水平的GAD65抗体,其次是脑脊液中也有GAD65抗体,同时还需证明该抗体是在鞘内合成。而本案例诊断为抗GAD65抗体相关性 CA,是因为血清与脑脊液中GAD65抗体为强阳性,高水平的GAD65抗体使神经系统疾病与GAD自身免疫性相关联[14]。
关于抗GAD65抗体综合征的治疗建议几乎都来源于小型临床试验,就目前现有研究表明,免疫治疗是该类疾病的主要治疗方法,未接受免疫治疗的患者大多出现较高的致残率[15]。2014年《美国医学会杂志》发表的一项回顾性研究指出,亚急性期的患者,激素、静脉注射用免疫球蛋白(IVIg)、血浆置换和利妥昔单抗可作为诱导治疗,后续维持期建议重复IVIg、长期口服激素或硫唑嘌呤等免疫抑制剂预防复发,且绝大多数患者远期预后良好;而慢性期患者,免疫治疗效果有限,复发率较高,且大多远期预后不佳[8]。由此可以断定,本类疾病的早期诊断及早期治疗极为重要,越早治疗,患者的预后可能就会越好。本文2例患者在明确诊断后立即给予免疫治疗,患者1因病程长达1 y,应用激素治疗后临床症状改善有限;患者2因急性起病入院,早期及时给予免疫球蛋白及激素治疗后患者症状改善较明显,预后较好。其后期疗效还需进一步随访观察。
综上所述,GAD抗体相关的自身免疫性神经系统疾病的临床症状多样,血清及脑脊液相关抗体的检测至关重要,临床上需要引起重视,以达到早期诊治的目的,免疫治疗是其主要的治疗手段。
