DNA/RNA提取及处理方法对评价沉积物中纤毛虫分子多样性的影响
2022-08-16李龙召黄平平徐奎栋
李龙召, 黄平平, 徐奎栋, 4, 赵 峰, 4
DNA/RNA提取及处理方法对评价沉积物中纤毛虫分子多样性的影响
李龙召1, 2, 黄平平3, 徐奎栋1, 2, 4, 赵 峰1, 2, 4
(1. 中国科学院海洋研究所海洋生物分类与系统演化实验室, 山东 青岛 266071; 2. 中国科学院大学, 北京 100049; 3. 山东省生物物理重点实验室, 德州学院生物物理研究院, 山东 德州 253023; 4. 中国科学院海洋大科学研究中心, 山东 青岛 266071)
环境DNA(eDNA)技术结合高通量测序已广泛应用于评价微型生物多样性与群落构成。相较于eDNA, RNA在环境中易降解, 环境RNA(eRNA)可更准确地反映群落近期生命活性状态。理论上, 基于eRNA检获的生物多样性要低于eDNA检获的总体多样性。但前期已发表研究显示同一站点eDNA获得的多样性低于eRNA检获量, 推测可能原因包括: 1)样本量不同; 2)RNA中存在DNA污染。为验证假设, 本研究以陆架区2个站位沉积物中的纤毛虫为实验对象, 系统性比较4种DNA/RNA提取方法: DNA直接提取(0.9 g沉积物), 大样本量DNA直接提取(2 g), DNA洗脱法(2 g), RNA直接提取法(2 g); 以及3种RNA提取后的处理方法: 无DNA酶反转录, 含DNA酶反转录, 先纯化RNA再反转录。研究结果表明, 大样本量(2 g)DNA直接提取法所检获的可操作分类单元(OTUs)约为小样本量(0.9 g)的2倍。相同样本量下, DNA洗脱法检获的OTUs最多, 而DNA直接提取检获的OTUs低于有/无DNA酶反转检获的数量, 先纯化再反转录所获得OTUs最少。就群落构成而言, DNA洗脱法和RNA纯化可有效降低浮游类群比例, 可更真实展现底栖群落的构成。综上, 建议使用DNA洗脱法评价沉积物中微型生物的总体多样性; 采用eRNA研究具有生命活性的生物群落时, 反转录之前可纯化RNA, 以获得更加准确的具有生命活性的群落信息。
eDNA; eRNA; DNA/RNA提取; 纤毛虫; 分子多样性
真核微生物包含原生生物, 小型后生生物和单细胞真菌等所有自养和异养类型[1-3]。海洋底栖真核微生物是食物网中的重要一环, 在物质循环和能量流动中发挥重要作用, 并维持着海洋生态系统的稳定[4-6]。纤毛虫作为海洋底栖真核微生物中重要一员, 作用广泛, 例如作为指示物种和遗传发育研究的模式生物[7], 以及稳定微食物网[8]等功能。研究海洋底栖纤毛虫多样性有利于深入了解生物地球化学循环过程[4-6, 8], 对认知海洋生态系统具有重要意义。
近年来, 环境DNA(eDNA)[9-11]和高通量测序技术的快速发展[12], 为研究纤毛虫等微型生物的分子多样性提供新的思路与方法。eDNA是指从环境样本中提取的总DNA, 无需分离目标生物[13]。eDNA在环境样本中存在时间长, 构成复杂, 来源广泛, 包含丰富的物种遗传信息, 既可检获具有活性的生物, 又可检获休眠状态生物以及来源于生物分泌物或者死亡个体的胞外DNA[14-15]。目前, 许多研究针对样本的空间异质性和反映生物群落的时效性, 以及实验流程的可重复性和稳健性展开探讨, 以期得到统一规范的实验流程[16-20]。基本实验流程包含: 采样及样品保存→DNA/RNA提取→引物设计, PCR扩增, 测序获得样本序列信息→数据分析, 获得生物群落信息[21]。目前, 提取方法的选用不尽相同, 已有多份研究探讨不同的DNA提取方法对分子多样性的影响[16, 22]。整体上, 试剂盒技术成熟, 在进行DNA提取时操作规范, 流程统一, 获得的DNA纯度高, 系统误差较小。目前大多数的分子研究都采用试剂盒提取[16-18, 22]。在使用试剂盒提取沉积物的研究中, 样本量和提取方法的选用争议较大[16, 18]。小的样本量(0.2~0.5 g)有利于机械化流程的开展[16], 而大样本量(>2 g)能更真实反映生物群落信息[23]。
相较于eDNA, RNA在环境中存在时间短[24], 作为细胞代谢的过程产物, 环境RNA(eRNA)能更为准确地反映群落活跃状态[25-26]。eRNA作为一种新技术, 尚缺乏一套标准化的方法来进行样本收集、处理和分析[27]。理论上, 同一样品, 基于eRNA检获的生物多样性要低于eDNA的检获量。然而, 课题组前期研究以及其他报道[16, 18]发现, 采用目前国际主流的DNA直接提取法(0.9 g沉积物)获得的多样性低于RNA方法(2 g沉积物)获得的多样性[28]。推测可能原因包括1)样本量不同; 2)在RNA反转录过程中, 污染基因组DNA的存在会对下游分析产生较大影响, 使得分子多样性结果偏高[29-30]。
为验证本研究提出的假设, 以陆架区2个站位沉积物中的纤毛虫为实验对象, 系统性比较4种DNA/RNA提取方法: DNA直接提取(0.9 g沉积物), 大样本量DNA直接提取(2 g), DNA洗脱法(2 g), RNA直接提取法(2 g); 以及3种RNA提取后的处理方法: 无DNA酶反转录, 含DNA酶反转录, 先纯化RNA再反转录。旨在评价: 1) 不同沉积物样本量对基于eDNA技术检获的纤毛虫分子多样性的影响。2) 相同样本量, 不同DNA/RNA提取方法对分子多样性评价的影响。3) 不同RNA处理方法对评估纤毛虫分子多样性的影响。以期提出利用eDNA/eRNA研究海洋沉积物中真核微生物的分子多样性的最佳研究策略。
1 实验方法
1.1 样品采集与样品保存
2018年8月搭乘“科学三号”科学考察船于黄海3600-3(35°39′86″N, 112°29′71″E)和3600-6站位(36°00′07″N, 123°00′02″E), 利用0.1 m2改进型的Gray-Ohara箱式采泥器, 进行沉积物样品采集。在每个站位采集含有上覆水的未受扰动的沉积物, 刮取0~2 cm表层沉积物约20 g放入封口袋中, 3600-3站位采集4个重复, 3600-6站位采集3个重复。置于–20 ℃超低温冰箱中保存, 以待后续DNA提取。
1.2 分子生物学方法
1.2.1 DNA/RNA提取
1) 取0.9 g沉积物, 采用Power Soil DNA Isolation Kit(Qiagen, Germany)进行DNA提取, 分3次进行, 每次0.3 g (DNA直接提取0.9 g); 2) 取2 g沉积物, 采用Power Max Soil DNA Isolation kit(Qiagen, Germany)进行DNA提取, 每个重复提取1份DNA (DNA直接提取2 g); 3) 取2 g沉积物, 采用Power Soil RNA Isolation Kit(Qiagen, Germany)提取RNA, 同时采用RNA Power Soil®DNA Elution Accessory Kit(Qiagen, Germany)于同一样品洗脱DNA, 每个重复提取1份RNA和1份DNA(DNA洗脱法2 g)。
1.2.2 RNA反转录
对提取的RNA采用3种不同的方式反转录处理获取cDNA: 1) 采用不含DNA酶的反转录试剂盒Prime Script II 1st strand cDNA Synthesis Kit(TaKaRa, Japan)直接反转录(无DNA酶反转); 2) 采用含DNA酶的反转录试剂盒5X All-In-One Master Mix (Gentaur, Europe)进行反转录(含DNA酶反转); 3) 先采用TURBO DNA-free™ Kit(Thermo Fisher, USA)对提取的RNA进行纯化, 纯化后的RNA采用Prime Script II 1st strand cDNA Synthesis Kit反转录(先纯化再反转)(图1)。
1.2.3 PCR与高通量测序
采用巢式PCR技术扩增纤毛虫的18S rRNA基因的V4高变区, 选用纤毛虫特异性引物CilF、Cil RⅠ、Cil RⅡ、Cil RⅢ[31], 以及真核微生物18S V4 高变区通用引物EukF、EukR[32]进行扩增, 每个样品设置3份PCR重复及1份阴性空白对照, 具体巢式PCR流程参考文献[33]。扩增后使用琼脂糖凝胶电泳检测PCR产物质量及片段长度(480 bp), 将同一个样品的3份PCR产物合并, 送至测序公司开展测序。
1.3 生物信息分析
测序数据进行拼接得到原始数据(raw data), 经过质量控制和去嵌合体得到可用于后续分析的有效数据(clean data)。质量控制包括: 1) 将Raw Tags从连续低质量值(≤19)碱基数达到设定长度(3)的第一个低质量碱基位点截断; 2) 进一步过滤掉其中连续高质量碱基长度小于Tags长度75%的Tags。对有效数据进行去冗余、去单一序列和 97%相似度的OUT (operational taxonomic units, 可操作分类单元)聚类分析, 根据 OTU 聚类结果, 对每个 OTU的代表序列与 SILVA 数据库比对作物种注释, 得到对应的物种信息和基于物种的丰度分布情况, 即OTU表格。利用EXCEL进行统计, 采用R进行单因素方差分析, 并绘制箱线图, 折线图, 韦恩图。使用PRIMER v6软件中的CLUSTER应用程序, 对同一样本量(2 g)的所有样本进行聚类分析。
2 实验结果
2.1 提取和处理方法对纤毛虫物种丰富度的影响
基于不同样品量的DNA直接提取法比较结果显示, 0.9 g样本量所获得的OTUs数明显低于2 g样本量所获得的OTUs数。在3600-3站位, 直接提取DNA: 0.9 g样本平均获得96个OTUs, 2 g样本量每个测序样本平均获得152个OTUs。在3600-6站位, 直接提取DNA: 0.9 g样本量平均获得83个OTUs, 2 g样本量每个测序样本平均获得161个OTUs。
在相同样本量(2 g)的情况下, 以3600-3站位为研究对象, 结果表明: 采用DNA洗脱法平均检获的OTUs最高; 其他方法排序: 依次为无DNA酶反转, 含DNA酶反转, DNA直接提取以及RNA先纯化再反转的方法。以3600-6站位为研究对象, DNA洗脱法平均所检获的OTUs亦最高; 其他依次为含DNA酶反转、DNA直接提取、无DNA酶的反转以及RNA先纯化再反转的方法(图2)。在3600-3站位, 不同提取方法下的单因素方差分析(方差齐性检验= 0.3, 符合正态分布)显示, 不同提取方法之间存在显著差异(= 0.023); 在3600-6站位, 不同提取方法下的单因素方差分析(方差齐性检验= 0.1, 符合正态分布)显示, 不同提取方法之间存在极显著差异(= 0.007 5)。

图2 3600-3与3600-6站位同一样本量不同提取处理方法所获得的OTU数量
2.2 提取处理方法与纤毛虫生物群落结构的关系
总体来看, 直接DNA提取的方法获得大量的浮游类纤毛虫(Choreotrichia 以及Oligotrichia)序列。采用样本量0.9 g, 在3600-3和3600-6站位平均获得的浮游类纤毛虫相对丰度最高(52%±0.155%; 51%±0.079%); 采用样本量2 g, 平均获得的浮游类纤毛虫相对丰度次之(41%±0.145%; 38%±0.157%); 其余的方法(DNA洗脱法与RNA提取方法)所获得的浮游类群相对丰度较低。在3600-3站位, 无DNA酶反转录方法获得的浮游类纤毛虫较高, 平均相对丰度为20.9%±0.116%, 其余依次为洗脱DNA方法(20.2%± 0.156%)、含DNA酶的反转录方法(13.9%±0.028%)以及RNA先纯化再反转方法(11.5%±0.076%)。在3600-6站位, 无DNA酶反转录方法获得的浮游类纤毛虫亦较高, 平均相对丰度为13.4%±0.073%, 其余依次为含DNA酶的反转录方法(7.9%±0.054%)、DNA洗脱方法(7.0%±0.015%)以及RNA先纯化再反转方法(1.5%±0.002%) (图3)。

图3 每个测序样本浮游类群(舞毛亚纲和寡毛亚纲)相对丰度折线图
在样本量相同的情况下, 综合所有重复, 分析不同方法获得的纤毛虫总体上以及删除浮游类群后所共有和特有的OTUs。结果表明, DNA洗脱法检获的特有OTUs数最多, 3600-3站位总体上51个, 删除浮游类群后47个; 3600-6站位总体上71个, 删除浮游类群后57个。而DNA提取法在两个站位检获的特异OTUs数波动较大(总体上, 3600-3: 6个; 3600-6: 33个)。有或无DNA酶反转所检获的特异OTUs数亦较高, 先纯化再反转所检获的特异OTUs数最低。以不同的RNA反转录方法为研究对象, 总体上看, 无论是在3600-3, 还是在3600-6站位; 含DNA酶反转录所获得的分子多样性和无DNA酶反转录所共检的OTUs比例较高, 均在50%以上。在3600-3站位, 总体上含DNA酶和无DNA酶共检获OTUs占比67.8%; 去除浮游类群后为65.6%。在3600-6站位, 总体上含DNA酶和无DNA酶共检获OTUs占比52%。去除浮游类群后发现为52.7%。综合两个站位, 对比含DNA酶反转和先纯化再反转的方法发现, 无论是否去除浮游类群, 先纯化再反转所检获的特异OTUs占比较少, 均在10%以下, 分别为6.9%, 7.8%, 2.9%, 3.3%(图4)。
分别对3600-3和3600-6站位相同样本量(2 g)的测序样本聚类分析, 结果显示: 在3600-3站位, 无DNA酶反转以及含DNA酶反转获得的纤毛虫群落结构较为相似, 然后与DNA洗脱法获得的样本群落聚合; 在3600-6站位, 含DNA酶反转获得的样本群落首先与DNA洗脱法获得的样本群落聚合, 其次与无DNA酶反转获得的样本群落聚合, 最后与DNA直接提取获得的样本群落聚合。结果显示, 无论是在哪个站位, 先纯化再反转所获得的样本群落信息与其他方法均在相似度50%以下聚合(图5)。
3 讨论
3.1 样本量对纤毛虫分子多样性的影响
研究表明, 不同样本量对重建土壤微生物群落(细菌, 真菌等)信息存在影响[34-36]。Nascimento等[18]利用不同的岩芯采样量评估了样本量对于真核生物多样性及群落结构的影响, 分析得出随着样本量的增加, 群落结构信息随之稳定; 而在Pearman等[16]的研究中发现在低重复样品数的情况下, 不同样本量检获的群落差异不显著。为降低沉积物样品异质性的影响, 我们的研究将每种处理方式设置3~4个重复, 结果显示不同样本量对纤毛虫分子多样性的评价具有重要影响。采用同种提取方法, 0.9 g样本量平均所获得的OTUs数约为2 g样本量所获得的OTUs数的1/2。从侧面证明样品量小是导致从0.9 g沉积物直接提取DNA获得的多样性低于从2 g沉积物提取RNA获得的多样性的重要因素。
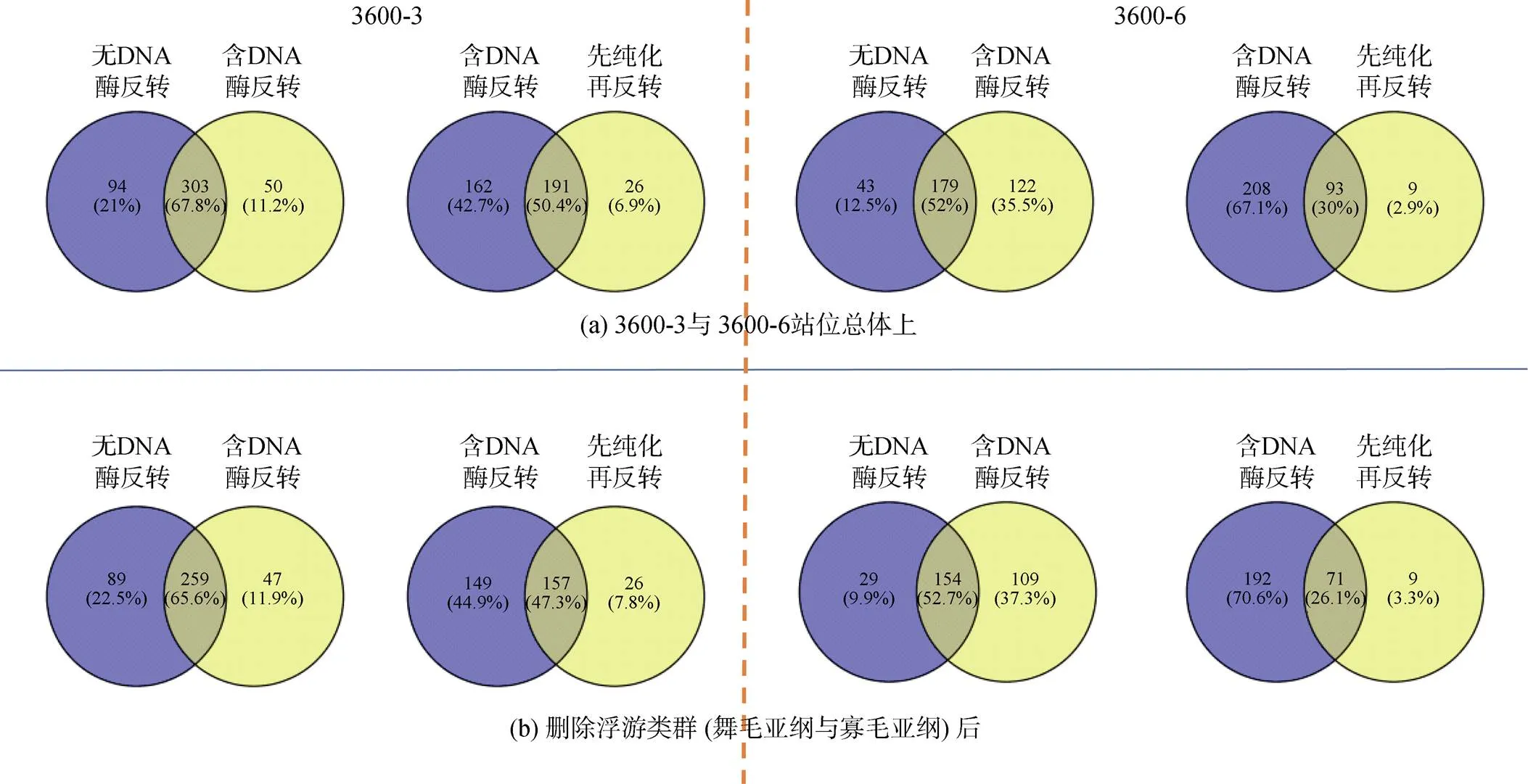
图4 3600-3与3600-6站位总体上和删除浮游类群(舞毛亚纲以及寡毛亚纲)后, 不同RNA处理方法之间共有和特有OTUs
生物群落构成分析结果显示0.9 g样本量所检测到的浮游类纤毛虫占比最多, 说明小样本量的DNA提取方法检测能力有限, 富集于海底的浮游物种的DNA会干扰底栖群落的研究。提取DNA时, 由于小样本量抽样不全[18], 会导致多样性的漏检, 进而形成生物群落构成的偏好性等。
3.2 不同提取方法对纤毛虫分子多样性的影响
利用eDNA作为物种检测手段已经十分成熟, 但目前仍没有一套统一规范的实验流程。现有的研究结果表明, 从环境样品中提取DNA, 不同的提取处理方法会导致分析结果的差异[16, 22]。本研究发现相较DNA提取法, DNA洗脱法可检获更多的OTUs, 这可能归因于直接提取DNA时, 腐殖质含量高, 更易与DNA的结合, 进而使得获得的生物遗传信息降低[37]。DNA洗脱法可更高效除去腐殖质的影响, 获得更为全面的分子多样性信息, 可为 eDNA研究提供可靠的模板[38]。
相较于eDNA, RNA在环境中存在时间短[24], 作为细胞代谢的过程产物, 环境RNA(eRNA)能更准确地反映群落近期的生命活性状态[25-26]。相较于有/无DNA酶反转录, 先纯化再反转录所获得的OTUs数量明显偏低; 有DNA酶反转录所获得的OTUs比无DNA酶反转录所获得的少, 推测可能是样本中残留的基因组DNA所致。这表明在RNA反转录过程中去除基因组污染是必要的[39]。
分析不同提取和处理方法所得群落信息, 发现采用RNA反转录方法所获得的浮游生物占比较少, 且纯化后RNA获得浮游生物占比最少。先纯化再反转录所获得的群落单独聚合, 并与其他方法之间存在差异(相似度小于50%)。考虑到RNA作为一种易降解的活性遗传物质[40], 我们推测先纯化再反转的方法可去除环境样本中较长时间内的基因组DNA等污染, 因此导致RNA先纯化再反转获得的OTUs也较少, 且群落构成与其他方法存在差异, 这也从侧面证实RNA先纯化再反转可更加准确的展现具有生命活性的生物群落。

图5 3600-3和3600-6站位相同样本量(2 g)测序样本聚类分析图
4 结论
增加样本量(0.9 g至2 g范围内)可检获更多的OTUs。相较于DNA提取法, DNA洗脱法可更加全面的反映微型生物的群落结构。RNA在环境样本中存在时间短, 可反映具有生命活性的微生物群落信息情况, 但直接提取的eRNA中存在DNA污染, 提取后直接反转录, 会检获较高比例的浮游类群。RNA提取纯化后再反转录可有效降低浮游类群比例, 可更加真实展现具有生命活性的底栖生物群落。综上, 我们推荐采用DNA洗脱法评价沉积物中微型生物的整体多样性; 采用eRNA研究具有生命活性的生物群落时, 反转录之前可先纯化RNA, 以获得更加准确的具有生命活性的底栖生物群落信息。
致谢: 中国科学院海洋研究所徐雨协助样品采集, 谨致谢忱。
[1] MCGRATH C L, KATZ L A. Genome diversity in microbial eukaryotes[J]. Trends in Ecology & Evolution, 2004, 19(1): 32-38.
[2] BIK H M, SUNG W, DELEY P, et al. Metagenetic community analysis of microbial eukaryotes illuminates biogeographic patterns in deep-sea and shallow water sediments[J]. Molecular Ecology Resources, 2012, 21(5): 1048-1059.
[3] 徐奎栋. 海洋微型底栖生物的多样性与地理分布[J]. 生物多样性, 2011, 19(6): 661-675.
XU Kuidong. Biodiversity and biogeography of marine microbenthos: progress and prospect[J]. Biodiversity Science, 2011, 19(6): 661-675.
[4] SHERR E B, SHERR B F. Significance of predation by protists in aquatic microbial food webs[J]. Antonie Van Leeuwenhoek International Journal of General and Molecular Microbiology, 2002, 81(1/4): 293-308.
[5] ARISTEGUI J, GAASOL J M, DUARTE C M, et al. Microbial oceanography of the dark ocean’s pelagic realm[J]. Limnology and Oceanography, 2009, 54(5): 1501-1529.
[6] SNELGROVE P, BLACKBURN T H, HUTCHINGS P A, et al. The importance of marine sediment biodiversity in ecosystem processes[J]. Ambio, 1997, 26(8): 578-583.
[7] PUCCIARELLI S, BALLARININ P, BARCHETTA S, et al. Ciliates as models to study diversities in microtubule functions by functional genomics[J]. New Biotechnology, 2010, 27: S32.
[8] 陈伊凡. 海洋纤毛虫摄食微型藻类过程中磷的动力学研究[D]. 厦门: 厦门大学, 2014.
Chen Yifan. Kinetics of phosphorus uptake by marine ciliates on microalgae[D]. Xiamen: Xiamen University, 2014.
[9] JERDE C L, MAHON A R, CHADDERTON W L, et al. “Sight-unseen” detection of rare aquatic species using environmental DNA[J]. Conservation Letters, 2011, 4(2): 150-157.
[10] DEJEAN T, VALENTINI A, DUPARC A, et al. Persistence of Environmental DNA in Freshwater Ecosystems[J]. PloS One, 2011, 6(8): e23398.
[11] CORINALDESI C, BARUCCA M, LUNA G M, et al. Preservation, origin and genetic imprint of extracellular DNA in permanently anoxic deep-sea sediments[J]. Molecular Ecology, 2011, 20(3): 642-654.
[12] LOMAN N J, MISRA R V, DALLMAN T J, et al. Performance comparison of benchtop high-throughput sequencing platforms[J]. Nature Biotechnology, 2012, 30(5): 434-439.
[13] TABERLET P, COISSAC E, HAJIBABAEI M, et al. Environmental DNA[J]. Molecular Ecology, 2012, 21(8): 1789-1793.
[14] HARRISON J B, SUNDAY J M, ROGERS S M. Predicting the fate of eDNA in the environment and implications for studying biodiversity[J]. Proceedings of the Royal Society B-Biological Sciences, 2019, 286(1915): 20191409.
[15] PIETRAMELLARA G, ASCHER J, BORGOGNI F, et al. Extracellular DNA in soil and sediment: fate and ecological relevance[J]. Biology and Fertility of Soils, 2009, 45(3): 219-235.
[16] PEARMAN J K, KEELEY N B, WOOD S A, et al. Comparing sediment DNA extraction methods for assessing organic enrichment associated with marine aquaculture[J]. PeerJ, 2020, 8: e10231.
[17] GUERRIERI A, BONIN A, MUNKEMULLER T, et al. Effects of soil preservation for biodiversity monitoring using environmental DNA[J]. Molecular Ecology Resources, 2020, 30(13): 3313-3325.
[18] NASCIMENTO F J A, LALLIAS D, BIK H M, et al. Sample size effects on the assessment of eukaryotic diversity and community structure in aquatic sediments using high-throughput sequencing[J]. Scientific Reports, 2018, 8(1): 11737.
[19] CALDERONSANOU I, MUNKEMULLER T, BOYER F, et al. From environmental DNA sequences to ecological conclusions: How strong is the influence of methodological choices?[J]. Journal of Biogeography, 2019, 47(1): 193-206.
[20] PEREZBROCAL V, MAGNE F, RUIZ S, et al. Optimized DNA extraction and purification method for characterization of bacterial and fungal communities in lung tissue samples[J]. Scientific Reports, 2020, 10(1): 17377.
[21] ZINGER L, BONIN A, ALSOS I G, et al. DNA metabarcoding-Need for robust experimental designs to draw sound ecological conclusions[J]. Molecular Ecology, 2019, 28(8): 1857-1862.
[22] CHANGEY F, BLAUD A, PANDO A, et al. Monitoring soil microbial communities using molecular tools: DNA extraction methods may offset long-term management effects[J]. European Journal of Soil Science, 2020, 31(1): 134-148.
[23] LEAR G, DICKIE I, BANKS J, et al. Methods for the extraction, storage, amplification and sequencing of DNA from environmental samples[J]. New Zealand Journal of Ecology, 2018, 42(1): 10, 1A-50A.
[24] WOOD S A, BIESSY L, LATCHFORD J L, et al. Release and degradation of environmental DNA and RNA in a marine system[J]. Science of the Total Environment, 2020, 704: 135314.
[25] GUARDIOLA M, WANGENSTEEN O S, TABERLET P, et al. Spatiotemporal monitoring of deep-sea communities using metabarcoding of sediment DNA and RNA[J]. PeerJ, 2016, 4: e2807.
[26] PAWLOWSKI J, ESLING P, LEJZEROWICZ F, et al. Environmental monitoring through protist next-generation sequencing metabarcoding: assessing the impact of fish farming on benthic foraminifera communities[J]. Molecular Ecology Resources, 2014, 14(6): 1129-1140.
[27] WOOD S A, POCHON X, MING W, et al. Considerations for incorporating real-time PCR assays into routine marine biosecurity surveillance programmes: a case study targeting the Mediterranean fanworm (Sabella spallanzanii) and club tunicate (Styela clava)[J]. Genome, 2019, 62(3): 137-146.
[28] HUANG P, ZHAO F, XU K. Complementary DNA sequencing (cDNA): an effective approach for assessing the diversity and distribution of marine benthic ciliates along hydrographic gradients[J]. Journal of Oceanology and Limnology, 2020, 39(1): 208-222.
[29] LARMAN M G, KATZJAFFE M G, MCALLIE B, et al. Analysis of global gene expression following mouse blastocyst cryopreservation[J]. Human Reproduction, 2011, 26(10): 2672-2680.
[30] ALKHALIL A, HAMMAMIEH R, HARDICK J, et al. Gene expression profiling of monkeypox virus-infected cells reveals novel interfaces for host-virus interactions[J]. Virology Journal, 2010, 7: 173. DOI: 10.1186/ 1743-422X-7-173.
[31] LARA E, BERNEY C, HARMS H, et al. Cultivation-independent analysis reveals a shift in ciliate 18S rRNA gene diversity in a polycyclic aromatic hydrocarbon-polluted soil[J]. FEMS Microbiology Ecology, 2007, 62(3): 365-73.
[32] STOECK T, BASS D, NEBEL M, et al. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water[J]. Molecular Ecology, 2010, 19: 21-31.
[33] STOCK A, EDGCOMB V, ORSI W, et al. Evidence for isolated evolution of deep-sea ciliate communities through geological separation and environmental selection[J]. BMC Microbiology, 2013, 13: 150. DOI: 10.1186/1471-2180-13-150.
[34] KANG S, MILLS A L. The effect of sample size in studies of soil microbial community structure[J]. Journal of Microbiological Methods, 2006, 66(2): 242-250.
[35] ELLINGSOE P, JOHNSEN K. Influence of soil sample sizes on the assessment of bacterial community structure[J]. Soil Biology & Biochemistry, 2002, 34(11): 1701-1707.
[36] PENTON C R, GUPTA V S R, YU J, et al. Size matters: assessing optimum soil sample size for fungal and bacterial community structure analyses using high throughput sequencing of rRNA gene amplicons[J]. Frontiers in Microbiology, 2016, 7: 824. DOI: 10.3389/fmicb. 2016.00824.
[37] LAKAY F M, BOTHA A, PRIOR B A. Comparative analysis of environmental DNA extraction and purification methods from different humic acid-rich soils[J]. Journal of Applied Microbiology, 2007, 102(1): 265- 273.
[38] LEJZEROWICZ F, ESLING P, PILLET L, et al. High- throughput sequencing and morphology perform equally well for benthic monitoring of marine ecosystems[J]. Scientific Reports, 2015, 5: 13932. DOI: https://doi.org/ 10.1038/srep13932.
[39] NORHAZLIN J, NORASHIKIN M N, HOH B P, et al. Effect of DNase treatment on RNA extraction from preimplantation murine embryos[J]. Genetics and Molecular Research, 2015, 14(3): 10172-10184.
[40] LI Y F, BREAKER R R. Kinetics of RNA degradation by specific base catalysis of transesterification involving the 2'-hydroxyl group[J]. Journal of the American Chemical Society, 1999, 121(23): 5364-5372.
Effects of DNA and RNA extraction methods for the evaluation of ciliate diversity in marine sediments
LI Long-zhao1, 2, HUANG Ping-ping3, XU Kui-dong1, 2, 4, ZHAO Feng1, 2, 4
(1. Department of Marine Organism Taxonomy and Phylogeny Institute of Oceanology, Chinese Academy of Sciences, Qingdao 266071, China; 2. University of Chinese Academy of Sciences, Beijing 100049, China; 3. Shandong Key Laboratory of Biophysics, Institute of Biophysics, Dezhou University, Dezhou 253023, China; 4. Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao 266071, China)
Environmental DNA (eDNA) has been used along with high-throughput sequencing to evaluate microbial diversity and community composition. Compared with eDNA, environmental RNA (eRNA) degrades easily and can be used to investigate the community of active taxon. The species richness obtained by eRNA is lower than that by eDNA in theory. However, previous studies have shown that the diversity obtained by eDNA at the same site is lower than that detected by eRNA. This unexpected finding may be attributed to the difference in the sample size and the DNA contamination in RNA. To test this hypothesis, we estimated the effects of DNA and RNA extraction methods on ciliate diversity in sediments obtained from the continental shelf area. Four extraction methods were performed: DNA direct extraction with 0.9 g sediments, DNA direct extraction with 2 g sediments, DNA elution, and RNA direct extraction from 2 g sediments. Meanwhile, three types of eRNA treatments before the reverse transcription, including the addition of DNA enzyme, no addition of DNA enzyme, and purifying RNA, were also compared. The results revealed that the number of operational taxonomic units (OTUs) detected from 2 g sediments was about twice that from the 0.9 g sediments by DNA direct extraction. Using the same sample size, the highest number of OTUs was detected via DNA elution and the lowest number by purified RNA. The number of OTUs detected by DNA direct extraction was lower than that by eRNA, irrespective of the addition of DNA enzyme. In terms of community composition, DNA elution and RNA purification can effectively reduce the proportion of planktonic groups, thereby improving the estimates of the composition of the benthic community. In summary, the DNA elution method is recommended for evaluating the total diversity of microorganisms in marine sediments. eRNA can be used after purification to investigate active communities.
eDNA: eRNA; DNA / RNA extraction; ciliate; molecular diversity
Sep. 22, 2021
[National Natural Science Foundation of China, No. 41876171]
Q958
A
1000-3096(2022)07-0052-09
10.11759/hykx20210922001
2021-09-22;
2021-10-28
国家自然科学基金资助项目(41876171)
李龙召(1997—), 男, 安徽定远人, 硕士研究生, 主要从事海洋真核微生物多样性研究, E-mail: 15375544229@163.com; 赵峰(1987—),通信作者, 副研究员, E-mail: fzhao@qdio.ac.cn
(本文编辑: 赵卫红)
