部分型雄激素不敏感综合征一例
2022-02-16李婕一杨雁胡蜀红余学锋何文涛
李婕一 杨雁 胡蜀红 余学锋 何文涛
患者,男,24岁,因“尿道下裂24年,双侧乳房发育9年”于2019年4月入院。患者24年前出生时即发现尿道下裂,伴右侧隐睾;22年前(2岁)行尿道下裂修复术;12年前(12岁)行右侧隐睾固定术,术后1年出现右侧睾丸萎缩;9年前(15岁)开始青春期发育,并发育迟缓,表现为阴茎短小,勃起长度约为5~6 cm,精液量少,伴胡须少、腋毛少、体毛少、喉结不突出。后逐渐出现双侧乳房发育,无头痛、视野缺损、泌乳症状。6年前曾至当地医院行双乳房切除术。患者现为进一步明确诊断至本院就诊。既往史:否认其他疾病病史;其父母青春期发育均无明显异常。体格检查:T 36.2 ℃,P 75次/分,R 20次/分,Bp 121/64 mmHg。身高160 cm,上部量80 cm,下部量80 cm,腹围75 cm,体重55 kg,BMI 21.48 kg/m2;心、肺、腹检查未见异常;专科体格检查:喉结不明显,腋毛稀少,胡须稀少;乳头较大,呈女性型发育;右侧腹股沟可见一长约10 cm陈旧性手术瘢痕,阴毛呈倒三角分布,Tanner 4期;睾丸椭圆形,右侧睾丸容积2 ml,左侧睾丸容积12 ml,阴茎牵拉长度约2.5 cm,尿道开口于阴茎头部下方。实验室检查:肝肾功能、血糖、甲状腺功能、皮质醇水平均正常。促卵泡激素(FSH)17.33 mIU/ml(1.27~19.26 mIU/ml,括号内为成年男性正常参考值范围,以下相同),黄体生成素(LH) 27.89 mIU/ml(1.24~8.62 mIU/ml),睾酮(T)5.42 ng/ml(1.75~7.81 mIU/ml),游离睾酮0.312 nmol/L(0.11~0.66 nmol/L),生物活性睾酮 7.31 nmol/L(2.80~15.50 nmol/L),游离睾酮指数 38.9%(24.3%~110.2%);雌二醇(E2)33 pg/ml(<47 pg/ml),孕酮(P) 1.57 ng/ml(0.10~2.06 ng/ml),性激素结合球蛋白(SHBG) 48.4 nmol/L(13.3~89.5 nmol/L),泌乳素(PRL) 14.65 ng/ml(2.64~13.13 ng/ml);外生殖器超声检查示:右侧睾丸体积小(1.4 cm×0.6 cm)并实质回声不均(发育异常);左侧睾丸实质回声尚均(4.6 cm×1.5 cm);双侧附睾形态大小正常,精索静脉未见明显迂曲扩张;盆腔磁共振成像(MRI)检查示:前列腺信号欠佳,精囊显示欠清,建议行前列腺MRI进一步检查;右侧睾丸萎缩,左侧阴囊内见结节状异常信号灶;垂体MRI增强检查未见明显异常。染色体核型分析:46,XY(小Y)。基因检测结果:患者存在一处基因突变:雄激素受体(AR)基因第5外显子c.2216A>G(腺嘌呤→鸟嘌呤),导致氨基酸改变p.Gln739Arg(谷氨酰胺→精氨酸)(图1)。总结患者的病情特点为:存在先天性尿道下裂,右侧隐睾,青春期发育缓慢,存在乳腺发育、胡须稀少和喉结不明显等雄性化不足,但实验室检验提示睾酮呈中高水平,且其上游调控激素LH水平明显升高。该患者并不存在雄激素水平的缺乏,而是存在雄激素作用的缺陷。同时,该患者无高血压,皮质醇水平正常,不支持先天性肾上腺增生疾病引起的男性假两性畸形;无长期药物服用史,排除药物所致乳腺发育的可能。鉴于此,我们对该患者进行了雄激素受体(AR)的基因检测,结果证实,该患者确实存在AR基因的突变。部分型雄激素不敏感综合征(PAIS)患者可表现为尿道下裂、隐睾症、青春期后男性乳腺发育等特征性表现,LH升高,T正常或升高,检测AR存在基因突变或功能异常。患者诊断为PAIS,给予双氢睾酮(安特尔)40 mg每日2次口服补充雄激素治疗;另告知其存在一定睾丸恶变风险,建议密切监测睾丸情况。患者1个月后随诊复查,诉性欲较前提升,体格检查见腋毛较前稍增多,复测FSH 15.38 mIU/ml,LH 30.86 mIU/ml,T 6.88 ng/ml,均较治疗前升高;仍需进一步随诊观察疗效并调整雄激素用药方案。
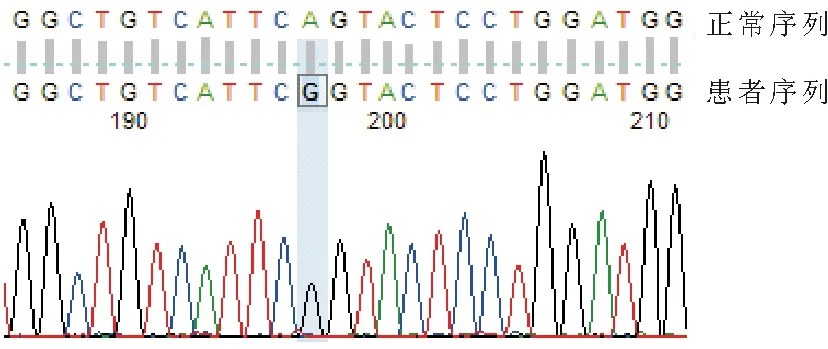
图1 患者外周血Sanger测序法AR外显子测序结果:第5外显子错义突变c.2216A>G;p.Gln739Arg
讨 论
雄激素不敏感综合征(AIS)是由于AR缺陷,导致靶细胞对雄激素反应能力低下或无反应的一类X连锁隐性遗传疾病。该病患者的染色体核型为46,XY,临床特点可表现为男性表型分化和(或)男性化异常。根据AR对雄激素敏感程度的不同,分为完全型AIS(CAIS)及PAIS及轻型AIS(MAIS)。
AIS的基本病理机制为雄激素反应能力的低下或缺失。雄激素发挥生物学作用需要AR及多种协同调节因子的作用[1]。AR属于类固醇受体,游离状态下存在于细胞浆内,与进入胞内的雄激素结合后形成激素-受体结合体,并在一系列辅助调控因子作用下进入细胞核,与DNA结合并进行转录调控,进而影响下游蛋白质合成,发挥雄激素的生物学效应。以上任一环节异常,均可导致雄激素作用通路的受损。当前已知,AR基因的突变是造成AIS的重要病因。AR基因位于Xq11-q12,由8个外显子组成。外显子1编码转录调节区,外显子2~3编码DNA结合区,外显子4~8编码铰链区和雄激素结合区。目前已有1 100余个AR突变类型被收录在数据库,其中较多数位于雄激素结合区及DNA结合区[2]。AIS可表现为表型多样性[3-4],有研究报道,同一致病家系可表现出CAIS及PAIS两种不同表型[5]。值得注意的是,AIS并非一定存在基因突变。相较CAIS中95%以上的AR突变率,PAIS的AR突变率仅为24%~43%[4-5]。对于无AR基因突变的PAIS,其发病机制尚在探究中。部分研究发现,无AR突变的PAIS患者,其出生体重相较有AR突变者降低,提示胎儿期生长受限可能与本病的发展相关[6-7]。另外,AR在发挥生物学作用时,可与>200个辅助调节因子互相作用[8],后者的异常亦可能对AR的作用产生影响。部分研究亦显示,雄激素受体功能异常的生殖器组织与正常组织相比存在不同的DNA甲基化模式[9-10],且AR基因近端启动子的CpG甲基化与其mRNA丰度明显呈负相关[11],提示表观遗传学在AIS的发病中存在一定作用。
AIS的临床表型取决于其雄激素的抵抗程度。正常男性胚胎在雄激素的作用下,其原始生殖结节增长形成阴茎,尿殖窦的下段伸入阴茎并开口于尿道沟,构成尿道海绵体部的大部分,两侧的尿生殖褶在中线融合形成阴茎缝。同时,雄激素促进胚胎中肾管发育为附睾、输精管和射精管等内生殖器,并促使睾丸下降。当雄激素作用不足时,以上过程受阻,临床上可表现为不同程度的去雄性化表现。PAIS患者的典型表现为出生时尿道下裂伴隐睾。因AIS不存在雄激素合成障碍,因此实验室检测睾酮水平表现为正常或偏高。青春期发育后,升高的睾酮可经芳香化作用转化为雌激素,继而出现男性乳腺发育症状。同时因睾酮对下丘脑-垂体系统的负反馈不足,LH水平通常明显高于正常男性,FSH亦可正常或轻度升高。临床中,根据典型的男性雄性化不足表现及高LH、正常/高T症状,PAIS较易与其他假两性畸形的病因的相鉴别,睾酮及LH水平是鉴别诊断中重要的一点。本例患者睾酮水平升高,可排除先天性睾酮合成障碍类疾病,如17β羟类固醇脱氢酶(17 β HSD)缺乏症、Leydig细胞发育不全等。患者染色体为46,XY,可排除染色体异常疾病如45,XO/46,XY嵌合体等。PAIS需要注意与5α还原酶缺陷症相鉴别,后者为睾酮向双氢睾酮转化障碍的一类疾病,亦表现为假两性畸形,睾酮水平不低,但表型多偏女性化,且其青春期后多无乳房发育,可作为鉴别的要点。因条件限制,本例患者未进行双氢睾酮水平检测,但其临床表现为典型的男性青春期乳房发育,且AR基因检测存在明确突变位点,因此可与5α还原酶缺陷症相鉴别。
本例患者存在X染色体中雄激素受体基因第5外显子第2 216位碱基已知点突变(2216A>G),造成所编码的第739位氨基酸由谷氨酰胺变为精氨酸(Q739R)。因患者家属拒绝配合检查,因此未对此位点进行家系验证。本例患者为独生子,自诉其父母及其他亲属无相似症状,且无性腺发育、生育障碍等问题。根据X连锁隐性遗传疾病的遗传特点,该突变可能来自于其母亲,理论上由母系携带并遗传;此外亦可能为本例患者的新发突变。该突变于2009年由Elfferich等[12]首次报道,该报道患者于4岁(青春期启动前)就诊,表现为小阴茎及尿道下裂,FSH 0.68 mIU/ml,LH 0.26 mIU/ml,T 0.1 nmol/L(HCG激发试验后20.5 nmol/L),与本例PAIS表型较一致。该患者行尿道下裂修补术后予局部DHT凝胶外用,3个月后阴茎增长1.6 cm,提示雄激素补充治疗有效。文献同时也对该突变其进行了转录活性、DNA结合能力、辅助调节因子结合能力等进行了测定,结果发现,本例突变可导致AR转录活性下降约50%,同时其与协同调节因子结合的能力基本消失。进一步提示了协同调节因子在AIS发病机制中的作用。
在临床表现中,PAIS通常在早期即发现明显的性发育异常,如尿道下裂、隐睾等。本例患者虽就诊较早,但未能及时明确诊断,导致青春期乳腺发育、阴茎发育不良。成年后,阴茎海绵体不再对雄激素有反应,即使通过雄激素替代治疗可改善部分第二性征及性欲等,但阴茎短小仍无法有效纠正,如能早期诊断,及时予以雄激素补充,可以避免青春期后的乳腺切除、阴茎短小。这提示在临床上,如遇泌尿外科尿道下裂、隐睾病史的儿童应排查AIS,以指导治疗。同时,AIS的另一重要表现为不孕不育。在CAIS患者中,因存在精原细胞缺乏及精子生成,恢复生育能力的可能性极小。PAIS患者尽管存在部分雄性化表现,但绝大部分仍无法生育。目前研究显示,由AR基因G824K及R840C突变引起的PAIS患者仍可能保留部分生育能力[13-14]。近期亦有AIS患者通过大剂量雄激素补充(庚酸睾酮,250 mg/周,维持4年)联合卵母细胞胞浆内单精子注射治疗后,恢复生育能力的报道[15]。
AIS的治疗需综合分析,多学科合作。对于PAIS患者,需尽早决定患者的社会性别,完成性别转化。研究发现,与CAIS患者相比,PAIS患者更需心理健康辅导。对于选择男性身份生活者,应采用尿道下裂修复术、乳房切除术等完成外形矫正;合并隐睾者应行隐睾固定术;同时根据雄激素抵抗程度,在青春期前及其后均应给予雄激素口服或注射补充治疗,用量取决于激素抵抗程度;局部宜应用DHT凝胶。研究发现,PAIS中睾丸癌变风险较CAIS高(>15%)[16],因此随访中需密切监测睾丸情况,必要时行睾丸活检判断有无原位癌。选择女性身份生活者,性腺切除在青春期前后均可。在青春期前切除者,需应用激素替代治疗诱发青春期(方案同Turner综合征),青春期后维持人工周期治疗;因青春期后睾丸癌变风险明显升高,在发育完成后尽早完成睾丸切除。本例患者口服安特尔1个月后复查,提示除T水平升高外,LH水平亦出现了轻度升高。可能存在以下几种原因:首先,安特尔剂量可能不足,对垂体的负反馈作用尚不明显,垂体在未接受到足够的负反馈信号情况下继续分泌LH;第二,垂体对激素的调节通常存在一定滞后反应,在雄激素对其产生负反馈后,短期内尚未能表现出激素的下调。本例患者虽未出现LH下调,但其随访时显示有部分临床症状改善,仍提示治疗有效,故建议其调整剂量,继续随访。
