假性甲状旁腺功能减退症家系临床特点及基因变异分析
2022-01-05桑艳红甄月巧李爽王楚楚
桑艳红 甄月巧 李爽 王楚楚
假性甲状旁腺功能减退症(PHP)是一组以终末靶器官对甲状旁腺激素(PTH)抵抗为特征,导致低钙血症、高磷血症和血清高PTH的一组罕见疾病[1]。该病主要是由于PTH与靶细胞膜受体结合异常及环磷酸腺苷(cAMP)依赖性通路活化受损而引起。PHP患者可能表现出典型的Albright遗传性骨营养不良(AHO)体征,即身材矮小、中心性肥胖、圆脸、颈短、短指/趾畸形,生化异常表现为低钙、高磷、高PTH及亚临床甲状腺功能减退症(SCH)。根据尿cAMP水平和对外源性PTH的反应性,PHP分为Ⅰ型和Ⅱ型。Ⅰ型主要表现为靶细胞膜受体-腺苷酸环化酶系统缺陷,对PTH不反应,不产生cAMP,PTH不能发挥激素的生理效应,尿cAMP降低甚至测不出,注射活性PTH后其尿cAMP和尿磷不增加;Ⅱ型则表现为PTH能够正常地与靶细胞膜受体结合产生cAMP,但cAMP不能进一步发生生理效应,尿cAMP升高、尿磷减少,注射活性PTH后尿cAMP继续上升,但尿磷不增加[2]。此外根据激素抵抗模式和是否具有AHO表型可将PHP Ⅰ型进一步分为3个不同的亚型:PHP-Ⅰa患者存在AHO表型和PTH、甲状腺素、促性腺激素、生长激素释放激素等多激素抵抗;PHP-Ⅰb具有典型的PTH抵抗特征,但没有AHO特征;PHP-Ⅰc患者的临床表现与PHP-Ia相似但G蛋白(Gsa)α亚单位的活性正常[3-4]。PHP-Ⅱ型与Ⅰb型类似,无多激素抵抗及AHO体型,补充外源性PTH后,cAMP生成无障碍,尿cAMP排出正常[5]。我们回顾性分析了1个Ⅰa型PHP患者家系的临床特征,通过基因检测确定疾病分型,旨在加强临床医师对该型PHP及该病基因检测的应用的认识。
对象与方法
1.对象:纳入先证者、先证者弟弟、先证者父母及外祖父母进行研究。本研究经郑州大学第五附属医院伦理委员会审批通过,患者及其家属均知情同意。
2.方法
(1)临床资料收集:收集先证者的一般信息、临床表现、体格检查、实验室检查资料。实验室检查资料包括:甲状腺功能、PTH、25羟维生素D3[25(OH)D3]、电解质、24 h尿钙、24 h尿磷、骨龄、促肾上腺皮质激素(ACTH)和皮质醇(COR)节律、胰岛素低血糖兴奋试验结果等。PHP、SCH诊断明确后,行全外显子基因测序;进一步完善先证者弟弟及父母亲的体格检查及相关实验室检查,最后获取先证者弟弟、先证者父母及先证者外祖父母的基因样本进行分析验证。
(2)基因检测:取先证者及其家属静脉外周血5 ml,采用血液基因组DNA Mini试剂盒(CwbioBiotech,北京,中国)从血液中分离出总DNA。使用Agilent Sure Select Human All Exon试剂盒进行捕获和文库构建,在Illumina平台上进行全外显子测序,有效测序数据与参考基因组(GRCh37/hg19)进行比对。结合基因变异频率、临床数据库(ClinVar、HGMD、OMIM)收录情况、软件预测结果(SIFT、MutationTaster、PROVEAN、MutationTaster等)筛选出一个GNAS基因的杂合变异c.534-2A>G,设计引物对筛选出的可疑候选变异进行PCR扩增和Sanger测序验证。PCR引物序列分别为:正向引物:5’-GGACGGTCACTTCCGTTGAG-3’,反向引物:5’-CGGTCACTCCACAAACCTGT-3’。
结 果
1.临床检查结果:先证者,女,9岁6个月,因“反复发作性上肢抽搐9年余,再发加重5天”于2014年4月3日入院。患者9年余前无明显诱因出现右上肢抽搐,右手腕部和肘部屈曲,持续约30 s自行缓解;此后上述症状间断发作4次,每次发作持续约30 s~2 min,可自行缓解;5天前再次出现双上肢紧张性抽搐,双下肢不明显,呼之不应,持续约3 min缓解。于我院门诊查PTH 946.8 pg/ml(14.0~72.0 pg/ml,括号内为正常参考值范围,以下相同),血钙1.34 mmol/L(2.03~2.54 mmol/L),血磷3.10 mmol/L(0.83~1.48 mmol/L)。体格检查:身高120 cm(同龄人身高中位数137 cm,-2 SD为125.0 cm),上部量60 cm,下部量60 cm,体重30 kg,BMI 20.8 kg/m2。脸圆面红、双手第4、5掌指关节较短,面神经叩击征(Chovestek征)阳性,束臂加压试验(Trouseau征)阳性,神经系统检查:腱反射亢进。实验室检查:血钙1.34 mmol/L,血磷3.41 mmol/L,血镁0.60 mmol/L(0.70~1.00 mmol/L),24 h尿磷12.06 mmol/L(22.00~48.00 mmol/L),24 h尿钙0.22 mmol/L(2.50~7.50 mmol/L)(尿量1 800 ml)。25(OH)D39.09 ng/ml(>20 ng/ml);碱性磷酸酶269 U/L(35~150 U/L),甲状腺功能五项:游离三碘甲腺原氨酸(FT3)6.24 pmol/L(2.80~6.80 pmol/L),游离甲状腺素(FT4)11.58 pmol/L(11.50~22.70 pmol/L),超敏促甲状腺激素(sTSH)12.734 mIU/L(0.500~5.500 mIU/L),甲状腺球蛋白抗体(TgAb) 16.9 U/ml(<60.0 U/ml),甲状腺过氧化物酶抗体(TPOAb) 20.3 U/ml(<60.0 U/ml))。性腺七项:总睾酮9.00 ng/dl,促卵泡生成激素4.92 mIU/ml,促黄体生成激素3.25 mIU/ml,泌乳素4.68 ng/ml,雌二醇26.20 pg/ml,孕酮0.373 ng/ml,生长激素0.596 ng/ml(0~8.000 ng/ml),提示垂体-性腺轴尚未启动。通过左手正位X线检查结果计算骨龄约11岁,大于实际年龄。胰岛素低血糖兴奋生长激素试验结果:生长激素0 min、30 min、60 min、90 min、120 min分别为6.92 ng/ml、2.86 ng/ml、0.588 ng/ml、0.232 ng/ml、0.674 ng/ml,高峰值>5 ng/ml但<10 ng/ml,提示垂体生长激素部分缺乏;患者早晨8时、下午4时、午夜12时的ACTH分别为28.9 pg/ml(7.2~63.3 ng/ml)、17.1 pg/ml(4.0~32.0 ng/ml)、3.3 pg/ml,COR分别为258.6 nmol/L(172.0~497.0 nmol/L)、142.2 nmol/L(74.1~286.0 nmol/L)、59.4 nmol/L。诊断考虑PHP、SCH。入院后予碳酸钙600 mg每日2次口服,骨化三醇(罗盖全)0.25 μg每日1次口服,优甲乐25 μg每日1次口服,复查血钙1.62 mmol/L,血磷2.88 mmol/L,血镁0.60 mmol/L。入院后患儿短暂发作1次抽搐,持续约30 s自行缓解。出院后长期予以钙剂、活性维生素D及优甲乐治疗,定期随访,未再发作抽搐。末次随访时间为2020年8月26日,患者身高140 cm,体重62 kg,BMI 31.6 kg/m2,FT34.46 pmol/L,FT413.48 pmol/L,TSH 7.44 mIU/L,PTH 261.8 pg/ml,血钙2.15 mmol/L,磷1.78 mmol/L,血镁0.70 mmol/L,25(OH)D323.4 ng/ml,将优甲乐剂量由原来每天50 μg调整为每天62.5 μg,罗盖全0.25 μg每日2次,钙尔奇D 300 mg每天3次。
先证者弟弟,男,4岁,外形、相貌与先证者相似,无明显临床症状。体格检查:身高104 cm,体重20 kg,BMI 18.5 kg/m2,脸圆面红,手指短粗,双手第4、5掌指关节较短,智力较同龄儿差。实验室检查结果:血钙2.24 mmol/L,血磷1.82 mmol/L,PTH 391.4 pg/ml,FT35.73 pmol/L,FT415.48 pmol/L,sTSH26.103 mIU/L。诊断为PHP、SCH。治疗上给予碳酸钙600 mg每日2次、罗盖全0.25 μg每日2次、优甲乐25 μg每日1次,长期随访。末次随访时间为2020年8月26日,患儿身高139 cm,体重47 kg,BMI 24.3 kg/m2,血钙2.15 mmol/L,血磷1.75 mmol/L,PTH 274.2 pg/ml,25(OH)D321.14 ng/ml,FT35.65 pmol/L,FT413.94 pmol/L,sTSH 6.726 mIU/L,目前身高在同年龄、同性别儿童中处于正常范围(约50%位置)。
先证者母亲,女,35岁,身高140 cm,体重56 kg,BMI 28.6 kg/m2,面圆颈短,十指粗短,面神经叩击征阳性。既往有低钙血症病史,间断补钙,检测血钙1.76 mmol/L,血磷1.54 mmol/L,PTH 174.3 pg/ml,FT34.78 pmol/L,FT412.75 pmol/L,sTSH 11.37 mIU/L,予钙剂、活性维生素D及优甲乐治疗。末次随访时间为2020年8月26日,复查血钙1.66 mmol/L,血磷1.50 mmol/L,血镁0.50 mmol/L,PTH 175.90 pg/ml,25(OH)D319.76 ng/ml,FT35.21 pmol/L,FT417.86 pmol/L,sTSH 1.745 mIU/L。
先证者外婆,女,65岁,身高138 cm,体重43 kg,BMI 22.6 kg/m2,患有糖尿病,体弱多病。
2.基因检测结果:我们在先证者外周血DNA样本的全外显子测序结果中发现了1个GNAS基因的杂合变异c.534-2A>G,通过Sanger测序对该变异位点进行验证,明确了先证者中该变异为杂合携带(图1)。随后对先证者家属进行该c.534-2A>G变异位点检测,结果显示,先证者母亲(Ⅱ:1)、外婆(Ⅰ:2)及弟弟(Ⅲ:2)均携带该变异位点,而在先证者父亲(Ⅱ:2)及外公(Ⅰ:1)未检出。本次研究检出的GNAS杂合变异c.534-2A>G在国内首次发现,该变异位于7号外显子相邻的经典剪切位置,且多种统计方法预测会对基因或基因产物造成有害影响。我们在GnomAD、ExAC、TOPMED数据库中对该变异频率进行检索,发现该变异在人群中的频率未见收录,不属于多态性变异。同时我们还发现先证者所携带的c.534-2A>G杂合变异已在一个有PHP表现的AHO家系中报道且与疾病共分离[6]。GNAS基因的经典剪切位点变化如c.435+1G>A、c.530+1G>T、c.973+2T>C、c.974-2A>G均已有PHP致病性的报道[7-8],该基因的剪切位点变异导致的PHP是已知的致病机理。根据ACMG指南判定该变异为致病性的。我们随后对该患病家系中其他成员中该变异位点进行验证结果显示先证者中该变异来自于其母亲,同时在先证者外婆及先证者弟弟中均检出该变异,而在先证者父亲及外公中未检出,提示在该患病家系中该变异由先证者外婆传递至其母亲,再由其母亲传递至先证者及其弟弟,该变异在该家系中属于母系遗传的基因失活突变引起的,按照基因诊断结果该患病家系为PHP-Ia型。
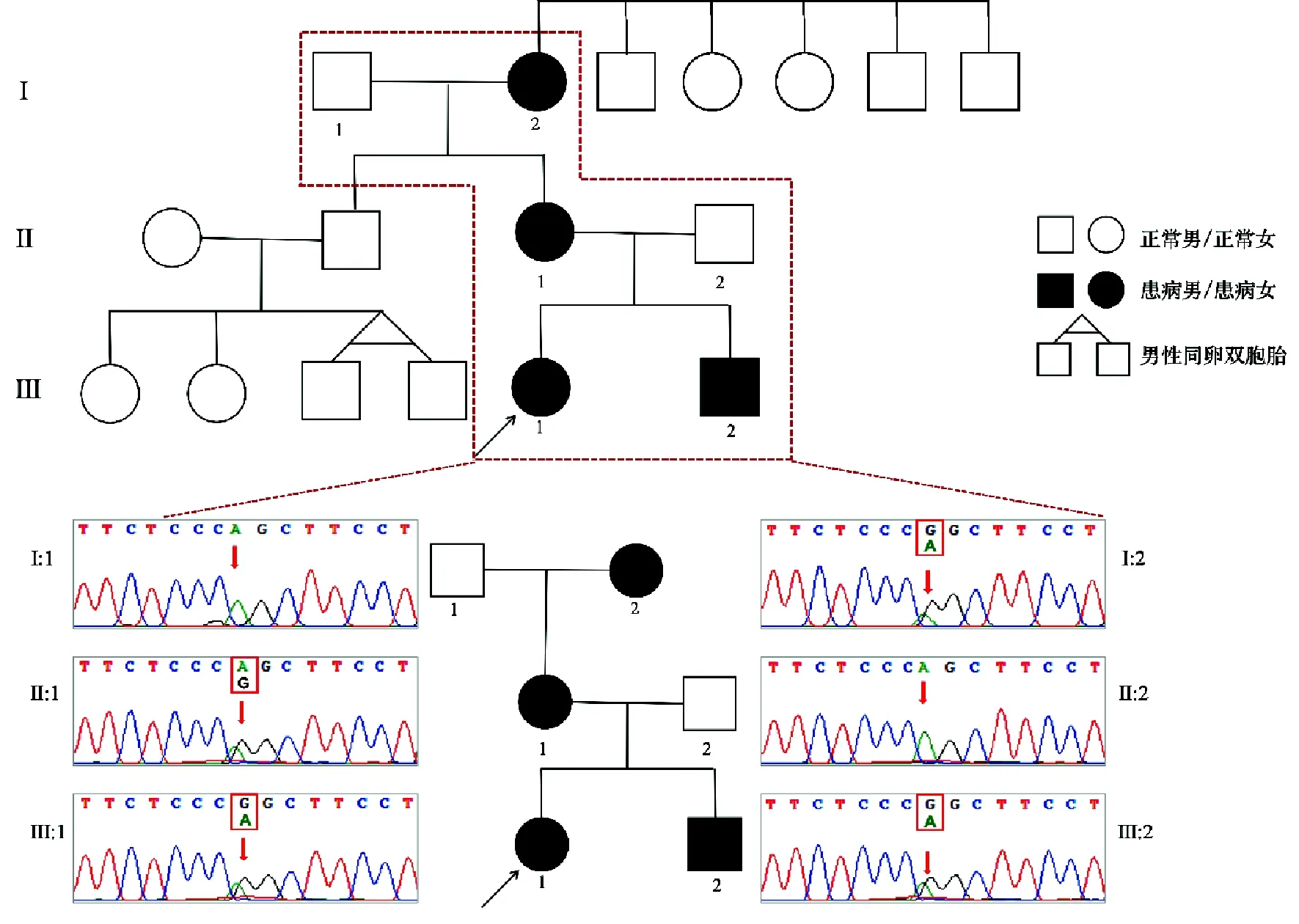
图1 研究对象家系图及GNAS基因变异Sanger测序结果:红色箭头表示变异位置:先证者(Ⅲ:1,黑色箭头所指)血液样本测序结果提示携带GNAS基因杂合变异c.534-2A>G;Ⅰ:1、Ⅱ:2未携带变异c.534-2A>G;Ⅰ:2、Ⅱ:1、Ⅲ:2携带GNAS基因杂合变异c.534-2A>G。同一位点出现双峰提示杂合。
讨 论
PHP是一种遗传性疾病,属于罕见的、高度异质性和严重损害的疾病,其特征是终末器官对PTH的作用产生抵抗[9]。PHP是Albright等描述的第一个激素抵抗综合征,且已证实与遗传有关,可由编码刺激性G蛋白(Gαs)α亚单位(Gsα)的GNAS外显子杂合失活突变引起[10]。已知在70%~80%的患者中,PHP-Ia是因为母系遗传的GNAS基因的杂合失活突变引起,而PHP-Ib通常为GNAS基因启动子区的胞嘧啶甲基化异常引起[11]。AHO表型与多种PHP有关,包括成人矮小、短肢骨化和皮下骨化。
GNAS基因定位于20q13.3,包含13个外显子,在结构上与小鼠2号染色体上的同源序列非常相似,具有高度复杂的印记表达模式,其能够调控父系表达的非编码RNA参与印记调节;主要编码刺激性Gsα,还可编码一个结构上不相关的蛋白Alex,参与配体互作、腺苷酸环化酶的激活及多种信号转导途径的调节。在啮齿类动物中的研究表明,Gsα的启动子和第一外显子位于1个富含CpG的岛上,但在这两个等位基因上都没有甲基化标记,这也是Gsα在大多数组织中是双向表达的原因[12]。然而,在肾近端小管、甲状腺、性腺、垂体、中枢神经系统的不同区域及棕色脂肪组织等少数组织中,Gsα主要来自母体等位基因,其父系等位基因通过尚未明确的机制被沉默,这也是母系遗传的基因失活突变引起的AHO会导致多激素抵抗、早发性肥胖和认知障碍的原因,而这种因母系突变导致的PHP被称为PHP-Ia型[13]。相反,由父系遗传的相同GNAS外显子失活突变引起的AHO不会引起激素抵抗、肥胖或认知障碍,因此被称为假假性甲状旁腺功能减退症(PPHP)。
结合我们收集的该家系临床信息,判断该家系符合PHP-Ⅰa型,主要表现为典型面容(圆脸、短颈)、身材矮小、低血钙、高血磷、高PTH,临床诊断为PHP、SCH。AHO会导致多激素抵抗、早发性肥胖和认知障碍。本研究中,先证者母亲的哥哥及其子女、先证者外婆的兄弟姐妹均无类似临床表现,结合之前的临床信息,该家系存在来源于母系的GNAS基因突变,在先证者诊断PHP后,其弟弟和母亲随后也发现除假性甲状旁腺功能减退之外,同时也并存SCH,并且这种功能减退是原发性SCH,而不是甲状腺激素抵抗,因为补充优甲乐后患者的sTSH可以恢复正常。值得注意的是,先证者诊断时其PTH水平明显升高,达正常值十几倍,低钙和高磷也更加明显,先证者母亲的低钙、高磷、高PTH水平次之,先证者弟弟的血钙正常,血磷、血PTH水平升高程度均明显低于先证者,提示即使是相同的基因突变,不同年龄患者的临床严重程度也并不完全一致。该家系经过6年的干预治疗后,先证者弟弟的生长发育影响较小,而先证者的甲状腺功能及钙磷代谢虽有所纠正,但终身高无明显改善,提示早期干预治疗可明显改善患者的临床结局。先证者母亲的低钙、高磷、高PTH与治疗前变化不大,考虑与其服药不规律有关。
对于PHP的治疗,因患者存在PTH抵抗,应给予活性维生素D治疗,但同时应积极给予普通维生素D的补充使其达到正常水平,以减少维生素D缺乏对其他组织器官的影响。
随着遗传学诊断技术的发展,越来越多的GNAS基因变异在PHP患者中被发现。本研究发现了与该家系呈疾病共分离的杂合变异c.534-2A>G,但尽管家系成员的临床信息与基因诊断结果均提示该家系为PHP-Ⅰa型,但我们没有对该变异进行功能学研究,还需更多研究来进一步评估这种独特的突变是否能导致Gsα的功能障碍。
