群体基因组结构变异检测工作流
2021-12-31曹舒淇刘诗琦
曹舒淇,刘诗琦,姜 涛
(哈尔滨工业大学 计算学部,哈尔滨150001)
基因组结构变异(Structural Variation,SV)是基因组上大尺度的核苷酸序列重排性变化,它包括长度大于50 bp的插入(INS)、缺失(DEL)、倒位(INV)、重复(DUP)、易位(BND)[1]。相关研究表明,平均每个人类个体上存在大约两万个结构变异[2],结构变异尽管相较于单核苷酸变异(SNV)、短插入缺失变异(INDEL)数量较少,但因其变异长度较大,因此对基因组上核苷酸序列的影响是最广泛的[3]。结构变异会改变基因序列信息,进而影响转录过程,改变蛋白质空间结构,从而引发性状与表型的改变[4]。此外,结构变异对基因表达调控[5]、种群多样性[6]等方面有着重要影响,同时与以自闭症[7]、阿尔兹海默症[8]等为代表的许多疾病的引发有密切的关系。
结构变异会对人类遗传、进化产生影响,形成个体之间的差异,影响种群的发展与演进。对于同一个群体,相当数量的结构变异对于群体中大部分个体是共享的,这些共享的结构变异可以有效对群体的特征与结构进行刻画[9]。此外,在群体中仍存在个体特有的结构变异,这些个体特有的结构变异反映了个体独有的特性,通过对特有结构变异以及个体表型的分析,能够发掘结构变异与表型、疾病之间的重要关系[10]。
随着国际千人基因组计划的实施与推动[11-13],各国也纷纷启动了本国的大规模人群基因组计划[14-17],希望通过分析和构建本国、本民族的基因组变异图谱,更加深入地解读本国人群在遗传、进化上的机理,为接下来开展的疾病诊治、精准健康发展提供支撑。结构变异作为对基因序列影响最为广泛的基因组变异类型,如何高效、精准的检测群体结构变异已成为当前群体基因组研究中的核心。因此我们基于多层过滤的质量控制,多种算法的联合检测、多维度变异融合和校对,开发了一个高性能的群体结构变异检测工作流,实现了群体基因组结构变异的全面、精准检测。该工作流总体分为四个环节:基因组测序片段比对,单样本基因组结构变异检测,单样本基因组结构变异融合以及群体基因组结构变异检测(见图1)。
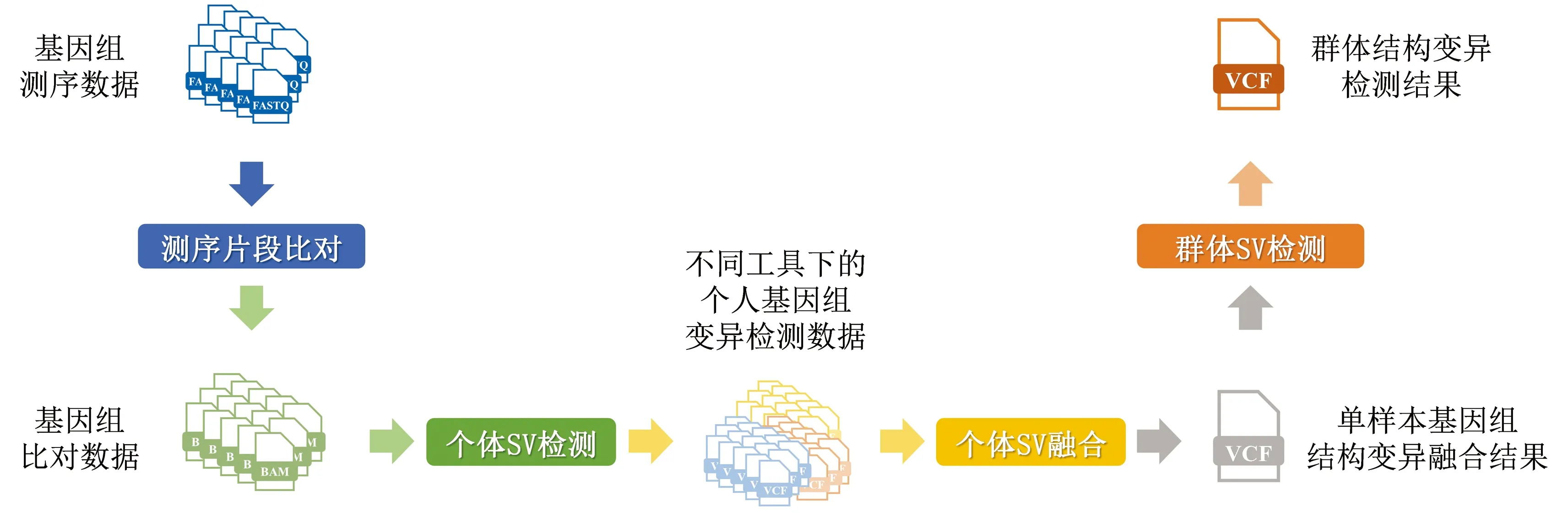
图1 群体基因组结构变异检测工作流Fig.1 Workflow of structural variation detection from population genomes
1 基因组测序片段比对
高通量基因组测序片段比对是基因组数据分析的首要环节,测序片段的比对的精度将对变异检测、基因组拼接等下游分析产生重要的影响。因此,对基因组片段测序数据、片段比对数据等有效的质量控制,是保障以测序片段比对为基础的基因组数据分析的关键。为此,本研究设计了多重质量控制与过滤的基因组测序片段比对工作流,该工作流主要包含以下步骤(见图2)。

图2 基因组测序片段比对流程Fig.2 Workflow of sequencing read alignment
(1)使用测序片段质量评价算法FastQC(https://github.com/s-andrews/FastQC)(v0.11.9,默认参数)对各样本基因组测序数据进行质量控制,通过对测序片段中GC含量、重复性、碱基质量、片段长度分布等指标进行统计和阈值判定,若任意满足:测序片段GC含量与理论分布偏差30%;测序片段重复度超过理论重复总量的50%;测序片段任意位置的碱基质量下四分位数低于5或中位数低于20;任意测序片段长度不足或长于150 bp,则将其认定为低质量测序样本数据,并进行过滤处理。
(2)使用高通量测序片段比对算法BWA (https://github.com/lh3/bwa)(v0.7.17,默认参数),完成各测序样本向参考基因组序列的比对。使用比对格式转换算法Sambamba[18](v0.8.0,默认参数)对各样本比对结果进行格式转换和排序;使用测序重复片段标记算法Samblaster[19](v0.1.2,默认参数)对各样本转换后的比对文件进行重复标记;使用GATK (https://github.com/broadinstitute/gatk)(v4.2.0.0,默认参数)对测序片段比对中碱基质量校正形成最终的片段比对数据。
(3)使用测序片段比对质量评价算法Qualimap[20](v2.2.1,默认参数)对各样本测序片段比对结果进行质量控制,通过对片段比对中测序覆盖度(不低于30×测序深度)、片段重复性(不高于5%)、片段比对率(不低于95%)等指标进行统计和阈值判定,进一步过滤低质量测序样本数据。
(4)使用DNA污染估计算法Verifybamid[21](v2.0.1,默认参数)计算各样本中DNA污染程度,过滤高污染率(高于3%)测序样本数据,形成最终用于下游分析的群体样本集合。
多重质量控制与过滤的基因组测序片段比对工作流在完成各样本测序片段比对任务的同时,将有效监控测序数据质量、比对数据质量、样本污染情况等多重指标,为高质量基因组结构变异检测奠定基础。
2 单样本基因组结构变异检测
受限于高通量测序数据读长与系统性测序误差的限制,采用单一结构变异检测工具识别各样本基因组中的结构变异往往存在敏感性与准确性较低的问题,这将制约结构变异的检测能力和向下游研究转化的水平。针对这一问题,本研究采用三款当前性能最好的个体基因组结构变异检测算法,全面挖掘各样本基因组结构变异,主要步骤如下(见图3)。

图3 单样本基因组结构变异检测流程Fig.3 Workflow of structural variation detection from individual sample
(1)使用快速检测基因组结构变异检测算法Manta[22](v1.6.0,默认参数)识别各基因组中DEL变异、INS变异、INV变异、BND变异、DUP变异。
(2)使用简化集成基因组结构变异检测与基因分型算法Smoove(https://github.com/brentp/smoove)(v0.2.7,默认参数)识别各基因组中DEL变异、INS变异、INV变异、BND变异、DUP变异。
(3)使用拷贝数变异检测算法CNVNator[23](v0.4.1,默认参数)识别各基因组中的拷贝数变异(CNV),并计算各基因组区域上测序覆盖度信息。
分别使用Manta、Smoove、CNVNator三种结构变异检测算法,将有效挖掘每个样本基因组中多种类型结构变异,为融合形成群体基因组结构变异提供支撑。
3 单样本基因组结构变异融合
群体基因组结构变异主要由各单样本基因组结构变异融合产生。如何对来自不同样本、不同检测算法形成的结构变异进行融合,是当前产生高精度群体基因组结构变异的核心。为此,本研究分别对群体样本中由同种检测算法与不同检测算法预测的结构变异分层次整合,从而产生最终群体结构变异候选位点,主要步骤如下(见图4)。
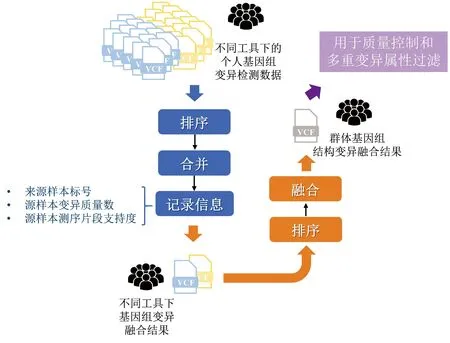
图4 单样本基因组结构变异融合流程Fig.4 Workflow of integration of individual structural variation
(1)分别对由Manta、Smoove检测算法产生的结构变异按照基因组坐标进行排序,完成在相同检测算法上不同样本结构变异的融合。若相邻两个结构变异存在交叠,则将两个变异合并为一个变异,直至所有变异均不存在交叠性。对于合并后的结构变异,分别记录来源样本标号,同时以累加方式累积各来源样本在此变异上的变异质量数、测序片段支持度等信息。
(2)再次对由Manta、Smoove检测算法产生的基因组结构变异融合结果进行排序,完成对不同检测算法产生的融合结构变异数据的二次融合。
通过使用不同工具对各样本基因组中的结构变异双重融合,在充分保留各样本基因组中潜在的结构变异的同时,在融合过程中记录每个结构变异的样本支持情况、变异质量情况等信息,为过滤低质量群体结构变异提供了保障。
4 群体基因组结构变异检测
在完成单样本基因组结构变异融合后,进行质量控制和多重变异属性过滤,是完成高质量群体基因组结构变异检测,绘制高精度人群基因组结构变异图谱的核心。本研究实现这一目标主要采用如下3个步骤(见图5)。
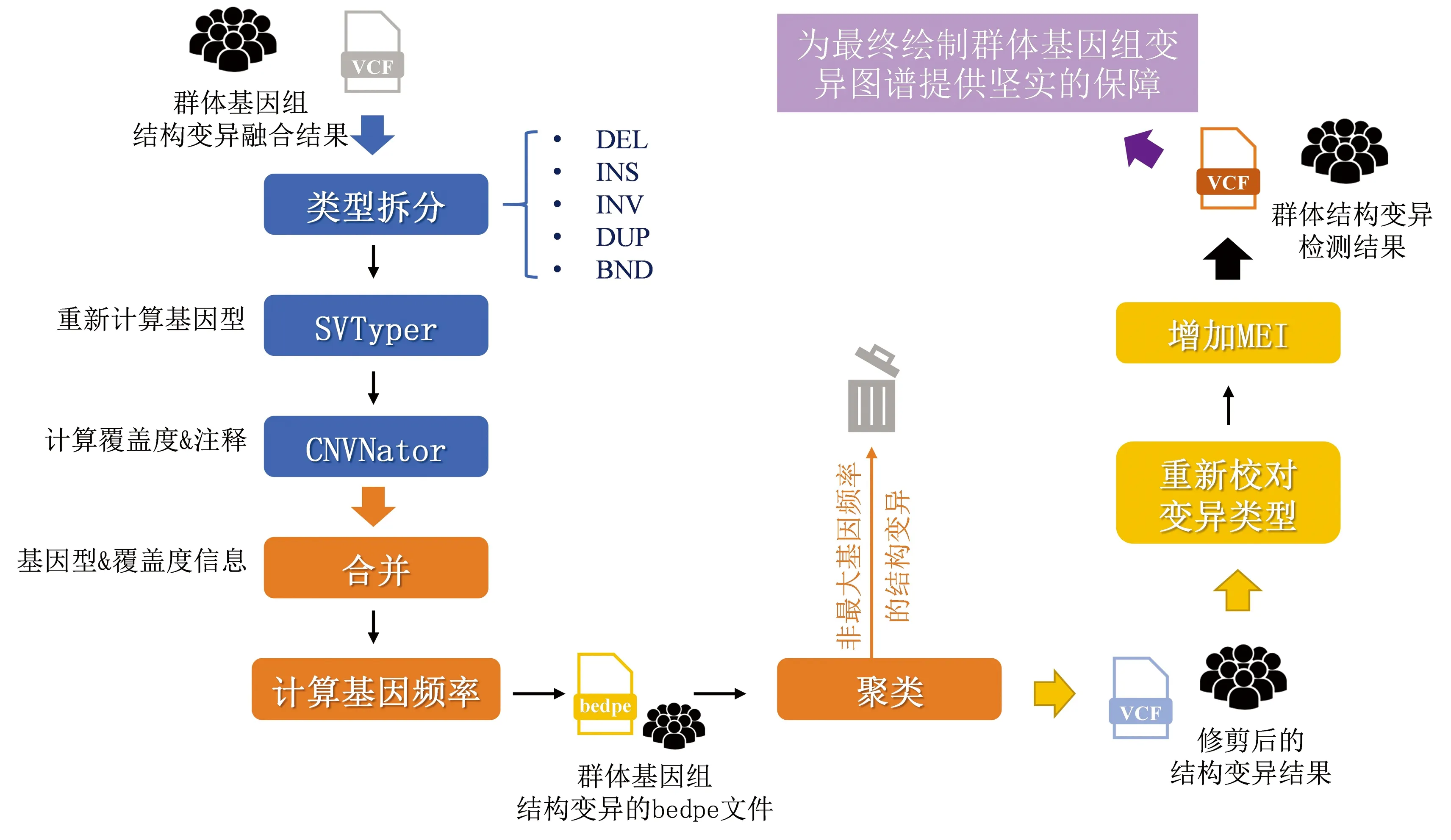
图5 群体基因组结构变异检测流程Fig.5 Workflow of structural variation detection from population genomes
(1)将融合后的基因组结构变异按照变异类型分别拆分为DEL、INS、INV、DUP、BND五种类型。使用基因组结构变异断点基因型计算算法SVTyper[24](v0.7.1,默认参数),分别从单样本层面对以上融合后的五种类型结构变异重新计算基因型。使用CNVNator计算的单样本层面的测序覆盖度信息对重新校准基因型信息的各结构变异进行注释。
(2)对重新校正基因型和测序覆盖度信息的各类型结构变异合并,计算各变异在人群中的变异频率。将合并后群体结构变异检测结果转换为bedpe文件格式并排序,对存在变异区域交叠的结构变异进行聚类,保留聚类中具有最大变异频率的结构变异,将聚类中其余结构变异修剪删除。
(3)将经过修剪的结构变异集合重新转换为vcf格式,依据测序覆盖度信息和基因型一致性信息对各结构变异重新校对变异类型,新增移动元件变异(MEI)类型,形成最终的群体结构变异检测结果。
经过对不同类型结构变异基因型的重新校正、过滤和变异类型校对,有效消减检测形成的假阳性结构变异预测结果,最大限度反映群体中真实的结构变异位点、类型和变异频率,为最终绘制群体基因组变异图谱提供了坚实的保障。
5 结果与分析
为了验证群体基因组结构变异检测工作流的真实效果,本研究构建了由267个样本组成的人群,使用Illumina高通量测序平台对该人群样本进行了30×高深度全基因组测序,并使用本研究提出的群体基因组结构变异检测工作流对此267个样本进行群体结构变异检测(见表1),合计检测出了96 202个结构变异,其中包括:11 697个DEL变异、18 385个INS变异、3 563个DUP变异、1 278个INV变异、2 007个MEI变异、59 272个BND变异。

表1 267样本人群中结构变异检测结果统计Table 1 Results of structural variation detection of 267 samples
在该267个样本构成的人群中(见图6),常见变异(AF≥0.05)占总体检出变异的41%(39 086/96 202),低频变异(0.05>AF≥0.01)占总体检出变异的18%(17 554/96 202),罕见变异(0.01>AF)占总体检出变异的41%(39 562/96 202)。值得关注的是,在DEL、DUP、INS、INV四种类型结构变异中,罕见变异的占比基本是均超过总体检验出变异的50%,相比之下,在MEI、BND两种类型结构变异中,检测出的常见变异数量是总体可检测变异数量的主要占比。这些结果与过去开展的基因组计划发现的结果相一致[25-26],说明本研究建立的群体基因组结构变异检测工作流具有良好的检测能力。

图6 不同变异频率中结构变异分布统计Fig.6 The distribution of structural variation among various allele frequencies
就每个样本可检测的结构变异而言,平均每个样本可以检测出18 388个结构变异,其中包含1 634个DEL变异、657个DUP变异、1 216个MEI变异、13 155个BND变异、1 521个INS变异、206个INV变异(见图7)。受限于高通量测序技术中读长的限制,基因组重复片段区域中的结构变异难以检测和精确分型,其中仅可检测到断点连接关系的结构变异均归结为BND变异,因此导致了每个样本中包含了相当数量的BND变异。然而,仅获取变异断点连接关系,无法解析结构变异精准结构(如:是否为平衡变异,DNA变化方向等)将严重影响BND变异的可信度和准确性。经过对BND变异按照置信度进行过滤(见图7),总计移除55 492个BND变异,仅保留3 780个高置信度BND变异(移除率93.62%)。平均每个样本移除11 703个BND变异,仅保留1 451个高置信度BND变异(移除率88.97%)。

图7 各样本不同类型结构变异检测数量分布和统计Fig.7 The quantitative distribution of various structural variation types among samples
此外,本研究还对群体基因组结构变异检测工作流中变异融合与群体变异检测两个关键环节的计算开销和内存使用进行了统计(见表2)。该工作流完成群体基因组结构变异检测和融合兼容串行分析和并行分析两种方式,其中串行计算方式需要约173.4 h,最大内存开销30 GB,而采用并行计算方式仅需不足3 h,并维持最大30 GB的内存开销。这一结果表明,对于大规模人群基因组结构变异检测分析,在保持有限内存消耗的前提下,采用并行方式运行该工作流将显著提升计算速度,为高效、快速的群体基因组结构变异检测提供了保证。

表2 267样本人群中结构变异检测运行时间及内存统计Table 2 Time and memory cost of structural variation detection of 267 samples
6 结 论
1)本研究构建了一套高效、精准的群体基因组结构变异检测工作流,该工作流通过多层过滤的质量控制,为高质量群体基因组结构变异检测提供支撑。
2)该工作流通过使用多种高性能结构变异检测算法,提高了结构变异检测的准确性与敏感性,并通过双重融合实现了群体结构变异候选位点的精准定位。
3)该工作流通过多维度重新校正结构变异候选位点的基因型与变异类别,进一步保障群体结构变异图谱的高质量构建。
4)利用该工作流对由267个样本组成的人群进行基因组结构变异检测,结果表明该工作流具有良好、快速、高效的检测能力。通过并行分析策略在控制内存消耗的基础上,提高了工作流的计算速度,为大规模群体基因组研究提供了可能。
