X连锁低磷酸盐血症性佝偻病家系1例报告并文献复习
2021-03-18
(青岛大学附属医院,山东 青岛 266003 1 内分泌代谢科; 2 内科学教研室; 3 医务科)
X连锁低磷酸盐血症性佝偻病(XLHR)一般始发于婴儿期,为X连锁显性遗传,是最常见的遗传性佝偻病。该病发病机制是由于磷酸盐调节基因(PHEX)突变影响了肾小管对磷的重吸收,从而导致尿磷排泄增多,血清磷酸盐水平下降。其临床特点主要表现为身材矮小,发育迟缓,下肢严重弯曲,如弓形腿,部分患者常伴有牙齿畸形。本病在临床上易误诊,传统治疗以口服磷酸盐补充剂联合活性维生素D类似物(阿法钙化醇或骨化三醇)为主,但是长期接受该疗法的成人患者容易引发甲状旁腺功能亢进和肾钙化。因此本文针对我院收治的一例XLHR家系的临床资料情况进行分析,并结合文献复习,探讨产前及产后的早期诊断对XLHR幼儿患者治疗及预后的意义。
1 临床资料
患者,女,36岁,自出生即检查出血清磷酸盐水平偏低。该患者幼时即有膝内翻表现,无其他伴随症状,曾接受过磷酸盐治疗,剂量未知。7岁时接受膝内翻矫形手术,术后不再服用磷酸盐,无其他不适。于2019年3月21日因左髋疼痛、久坐以及活动后疼痛加剧来我院就诊,患者左髋屈伸检查正常,后转诊至内分泌科。入院检查:血清碱性磷酸酶(ALP)117.00 U/L,血磷0.63 mmol/L,25羟维生素D3(25-OH-D3)20.00 μg/L,血钙2.16 mmol/L,甲状旁腺激素(PTH)1.55 μg/L,β-胶原降解产物0.22 μg/L,血肌酐35.00 μmol/L。腰椎正侧位、骨盆正侧位、左股骨正侧位X线检查显示骨盆骨密度降低,双侧骶髂关节、髋关节未见明显异常;左中股骨不规则,腰椎向右微弯曲。骨密度测定示腰椎T值0.62,左股骨T值-2.53。
患者儿子,2岁,12.6 kg。2019年2月1日于我院产科出生,其骨代谢相关指标检查结果显示,血磷为1.46 mmol/L,血钙为2.05 mmol/L,25-OH-D3为18.20 μg/L,PTH 0.51 μg/L,其他检查如血常规等未见明显异常。
患者母亲,58岁。2019年5月29号于我院门诊进行骨代谢相关指标检查示:血磷 0.67 mmol/L,血钙2.39 mmol/L,血肌酐60.00 μmol/L,血镁0.90 mmol/L,其他指标未见明显异常。
由于患者未进行产前遗传病检查,为明确诊断、指导治疗,于患者儿子出生2 d后同时采集患者及其儿子、母亲的血液标本进行骨骼发育异常相关基因检测,结果显示三者的磷酸盐调节内肽酶基因(PHEX)上均有一相同移码突变 (c.1097delT)(图1),该突变位点以前未见相关文献报道,因此初步诊断该患者家系为XLHR家系。
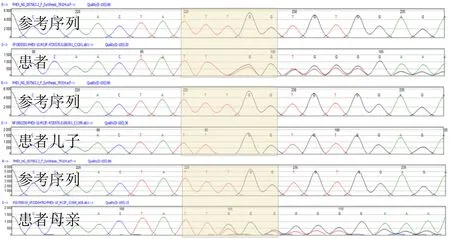
图1 患者及其儿子、母亲的PHEX基因突变位点图
基因检测后即给予患者儿子口服磷酸盐进行治疗,每日5次,每次10 mL;钙三醇丸每日1次,每次0.25 μg,患者未予药物治疗。1月后患者因左髋疼痛就诊后给予磷酸盐治疗,每日5次,每次30 mL;钙三醇丸每日1次,每次0.5 μg。患者母亲未予药物治疗。
3月后复查,患者血磷 0.76 mmol/L,25-OH-D323.50 μg/L,其他指标未有明显改善,自述髋部疼痛未有明显改善。患者儿子血钙2.18 mmol/L,其他指标未有明显改善,下肢X线检查显示下肢略弯曲,呈“O”形。患者母亲未接受复查。根据复查结果,将患者的治疗计划调整为磷酸盐每日5次,每次30 mL;钙三醇丸每日3次,每次0.25 μg。患者自述儿子无不良反应,故保持原治疗计划,并保持定期随访。
2 讨 论
XLHR是一种由肾脏磷酸盐消耗引起的低磷酸盐血症性疾病[1]。在临床上,XLHR的主要诊断依据是患者的发病年龄、临床症状、血清磷酸盐浓度是否降低、ALP浓度是否升高、PTH及尿钙是否正常,以及患者家系中是否存在有PHEX基因突变等[2]。本例患者幼年时即出现膝内翻,成年后以髋关节骨痛为主要症状,骨骼发育异常相关基因检测发现患者及其母亲、儿子均存在相同PHEX基因突变,故诊断为XLHR家系。
X连锁显性遗传是低磷酸盐血症性佝偻病最常见的可遗传形式,遗传发生率大约为1/20 000,约占HR的80%[2]。研究发现,XLHR与人类X染色体短臂上的PHEX基因的突变密切相关。PHEX基因位于人类染色体Xp22.1上,包含22个外显子,编码749个氨基酸。该基因主要表达于成骨细胞中[3],编码可降解局部整联蛋白结合配体、N-连接糖蛋白和骨桥蛋白的酶[4],还可以抑制血浆中磷脂酰肌醇和成纤维细胞生长因子(FGF23)的水平[5],PHEX基因突变可能导致内肽酶产生不足和肾脏磷酸盐排泄增加[6]。
目前关于XLHR的发病机制普遍认为是由于PHEX基因功能性突变,通过FGFR途径增加了FGF23的表达,从而影响肾小管对磷的重吸收[7-8]。另外,细胞外基质磷酸糖蛋白相关蛋白也可以影响FGF23的表达并调节肾脏磷酸盐的代谢[9]。还有研究表明,PHEX蛋白可以通过上调蛋白转化酶枯草杆菌蛋白酶/kexin-type2来裂解FGF23,并促进PHEX-人牙本质基质蛋白1-整合素复合物的形成,从而降低FGF23的水平[10],但是由于缺乏蛋白质之间直接相互作用的证据,这种说法目前仍然存在争议[11]。本例患者及其母亲、儿子的PHEX基因第10外显子中均发现有一处未见相关报道的突变(c.1097delT),该移码突变会导致其编码的蛋白质从第366位异亮氨酸开始发生移码并过早终止翻译,导致编码出的蛋白质截短体失去正常蛋白功能。尽管人类基因突变数据库已报告了400多个PHEX基因突变[12],但PHEX基因突变后所编码的蛋白尚不清楚,因此c.1097delT突变对XLHR的因果表型效应尚需进一步研究[13]。
XLHR在幼儿患者中很难做出诊断,极容易被误诊为维生素D缺乏症,只有当患儿出现生长发育迟缓、步态异常、颅骨畸形和骨痛等临床表现时,通过生化检查排除了维生素D缺乏症,再进行基因检测后才能确诊[14-15]。该患者儿子刚出生时骨代谢指标正常,依据骨代谢相关基因检测发现了PHEX基因突变,并参考其母亲及外祖母的基因检测结果,被确诊为XLHR。从生化角度看,XLHR患儿的特征是ALP升高,1,25-(OH)2-VitD3水平过低,血钙和25-OH-D3水平正常[16]。但是,新生儿正常的血清磷酸盐水平是成人的2倍,仅依靠骨代谢指标很难明确诊断,并且新生儿可有一定程度的足内翻,因此,患儿在2岁以内经常因忽视了骨骼畸形而错失治疗良机。此外,部分患儿常常以牙周脓肿为首发症状[17],因此部分父母可能会依据患儿的牙周症状误认为牙齿疾病,从而延误了XLHR的诊治。
由于大多数PHEX突变发生在87%的家庭病例和72%的散发病例中[18-19],因此仅依靠家族史很难推断出新生儿是否患有XLHR,只有通过基因检测并参考临床表型才能确诊。LIAO等[20]对2例患有XLHR的孕妇进行了基因检测,在1例患者家系中发现了一个新的错义突变c.1721T>A,其母亲和舅舅也有相同突变,通过从其羊水中提取的胎儿DNA测序显示,胎儿的基因位点在家族突变位点处为纯合子峰,初步判断胎儿出生后可能不会患有该病,婴儿出生后1个月进行基因检测证实其未患有XLHR;另一例孕妇患者产前未行胎儿基因检测,分娩后第2 天对新生儿进行基因检测,发现其携带与母体相同的突变,从而得到了及时治疗。
目前,XLHR的传统治疗方法主要是口服活性维生素D类似物(阿法钙化醇或骨化三醇)联合磷酸盐补充剂[17],但是用药剂量应根据血清中PTH、ALP和尿液中钙/肌酐的浓度随时调整,以降低严重副作用发生的风险[21]。临床研究表明,联合用药可以改善30%~60%的患者的骨痛症状[22],可以在1年内有效降低ALP浓度[17],减少由牙本质矿化等引起的牙周脓肿[23]。但需注意该疗法有引起甲状旁腺功能亢进和肾钙化的风险[24]。对于早期确诊的患儿,早期联合用药虽然有一定的效果,但仍然有24%~65%的患儿会出现下肢畸形,需要进行矫形手术[25]。
XLHR的新式疗法中研究最多的是应用人源化的FGF23单克隆抗体(Burosumab)进行治疗,其可通过抑制FGF23来改善磷酸盐转运蛋白的表达,从而提高患者血清磷酸盐水平[26]。在两项分别针对幼儿和成人的临床试验中已经证实,Burosumab可有效提高患者血清磷酸盐水平,且患者仅有轻至中度不良反应,如头痛及背痛等[27]。目前欧洲已经批准Burosumab为一种新的替代疗法,可用于欧洲1岁以上的患儿[14],但其是否会引起甲状旁腺功能亢进和肾钙化,尚不清楚,因此目前仍未被应用于常规治疗。
XLHR在疾病发展过程中易引起肾脏钙化、听力障碍、风湿病和心血管系统疾病等并发症,大大降低患者的生活质量。研究证实,接受早期治疗的患儿,身高标准差评分要优于出现骨骼症状后再接受治疗的患儿[28]。在出生后18个月内接受磷酸盐和骨化三醇联合治疗的患儿,相较于18个月内未经治疗的患儿生长速度有所提高[29]。因此,提倡对发现有XLHR家族史的患者进行婚前诊断,对有生育需求的患者进行产前的胎儿基因检测及产后的新生儿基因检测,这对患儿的早期诊断、治疗及预后都具有重要意义。
