SOTOS综合征NSD1基因新发错义突变1例并文献复习
2021-03-18
(青岛大学附属医院新生儿科,山东 青岛 266003)
SOTOS综合征为一种先天的过度生长性疾病,以常染色体显性遗传,主要表现为特殊面容、学习障碍以及生长过度,发病率约为1/14 000[1]。超过90%的患者存在NSD1基因异常,临床上症状与SOTOS综合征高度相似的非NSD1基因突变患者,已被证明携带APC2和NFIX基因突变[2]。本文报道1例NSD1基因新发错义突变患儿,并结合相关文献进行复习,探讨基因型与表型的关系,为提高临床医师诊断该病提供经验。
1 临床资料
对1例5月龄SOTOS综合征男婴的临床资料进行回顾性分析,并对患儿及父母进行全外显子组基因检测。获父母知情同意和医院伦理委员会批准后,采集患儿及父母外周静脉血各2 mL,EDTA抗凝,以QIAampDNA Blood Mini试剂盒(德国Qiagen GmbH公司)按厂家说明提取基因组DNA。将提取的基因组DNA片段化和扩增纯化以后与适配器相连接,由IDT XGen外显子研究小组(IDT,Lowa,美国)的探针捕获约19 396个靶向基因,构建DNA文库。DNA文库再经扩增和纯化以后,在NovaSeq 6000测序仪(Illumina,San Diego,美国)上采用Burrows-Wheeler Aligner(v.0.5.9-r16)软件进行测序,测序结果与UCSChg19人类参考基因组序列进行比对,使用PriVar Toolkit工具进行数据注释后,确定有临床意义的变异体,再用PCR方法验证检测到的变异,并在3500XL遗传分析仪上对获得产物进行直接测序。通过SIFT、PROVEAN PREDICTION、Mutation Taster、Polyphen2等生物信息学软件预测致病性。
2 结 果
2.1 临床表现及体征
患儿,男,5月龄,因“发育迟滞”就诊。系第2胎第2产,胎龄40+4周,顺产出生,出生时体质量为3 990 g,Apgar评分10分,无窒息史。母亲孕期体健。自出生以来,喂养困难,吮吸差,哭声直,现仍不能逗笑,不会咿呀作语,不能追视。出生后10 d因“新生儿高胆红素血症、新生儿肺炎”就诊当地医院,最高时总胆红素为389.9 μmol/L,住院10 d好转出院。家族史:父母体健,否认为近亲结婚,否认有家族遗传病史,患儿有1个3岁姐姐,体健。查体:头围49.2 cm,身高71 cm,体质量7 kg。前囟未闭,约2 cm×2 cm大小,平软。巨颅,颅骨重叠,前额突出,下颌尖长,高腭弓,下牙龈凹凸不平(图1A、B),萌牙2颗。患儿双耳廓外形基本正常,双耳(长约6 cm)、双手(长约8 cm)、双脚(长约11 cm)较同龄儿明显偏大,双侧指掌纹无异常,心、肺、腹查体未见异常。双手拇指内收,四肢肌张力高,病理征及脑膜刺激征阴性。抬头差,竖抱头不稳,视听追视无反应,不会逗笑,不能与人互动,哭声直,发音单一,不会咿呀作语,无主动抓握意识,不可独坐。精细运动、大运动及语言发育均落后同龄儿。
2.2 实验室检查结果
血常规、生化、心脏以及泌尿系统超声检查均未见有明显异常;颅脑MRI检查显示前后径增大,脑沟深,脑回增宽,双侧基底节区T1WI稍高信号,双侧内囊后肢T1高信号,双侧脑室体部异常信号(图1C~E)。
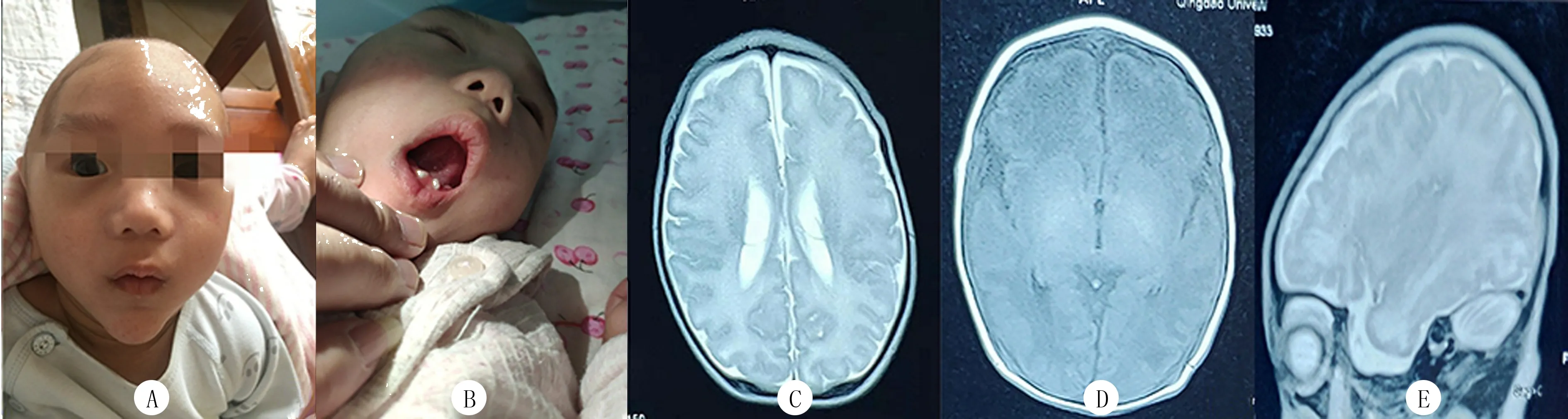
A:患儿的前额突起,下颌尖长;B:患儿下牙龈凹凸不平,牙列不齐;C~E分别为T1轴位、T2轴位、T1矢状位颅脑MRI图像
2.3 基因检测结果
基因检测结果显示,该患儿5号染色体5q35区出现NSD1基因区域错义突变c.5791T>C,使该位点的半胱甘酸转变为精氨酸(p.Cys1931Arg)。在HGMD数据库中并未见相关文献报道;dbSNP147数据库、ESP6500siv2_ALL数据库和千人基因(1000g2015aug_ALL)数据库均未见收录。其父母样本中未检测到该变异,因此推测其为新发突变。患儿、患儿父亲、患儿母亲的基因检测结果见图2。
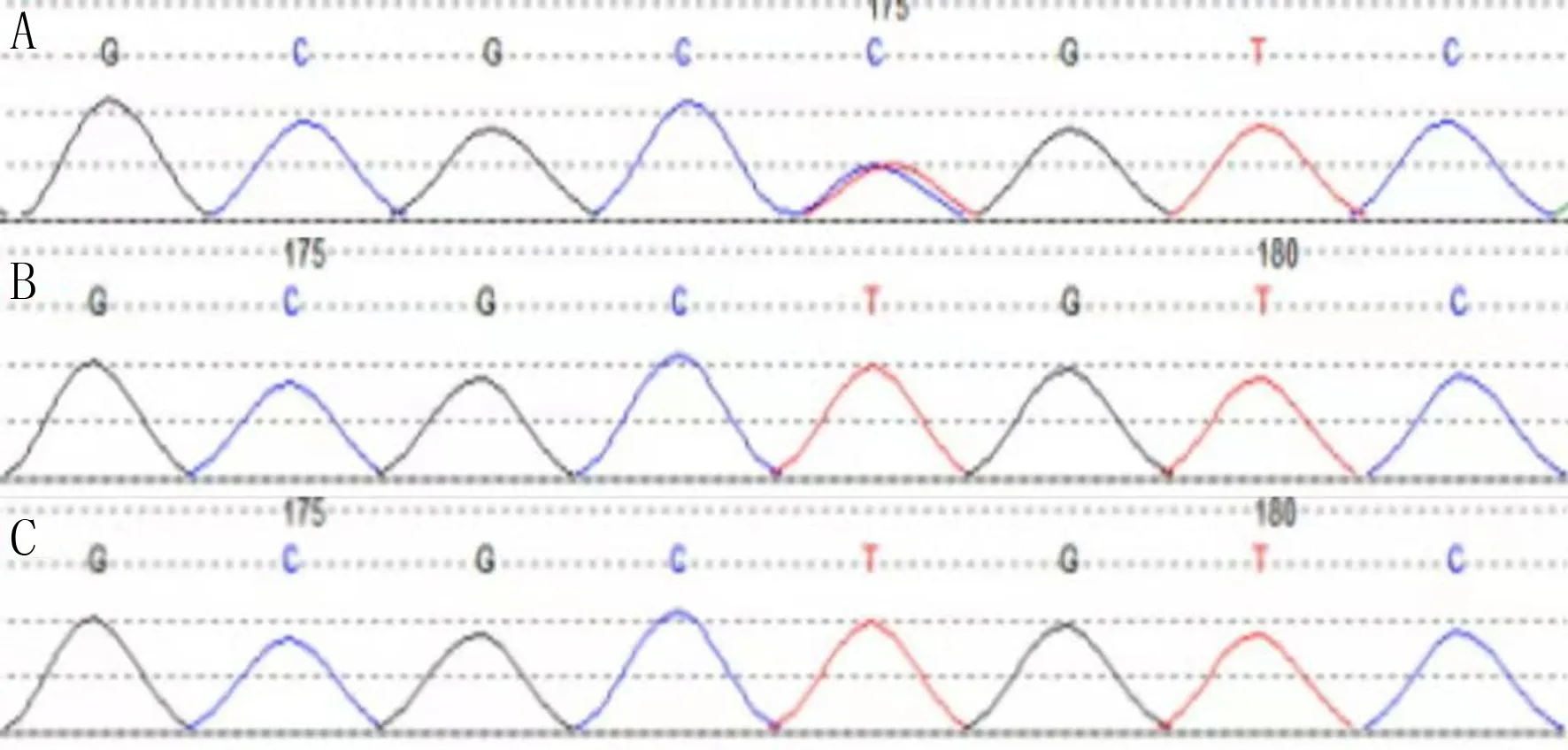
A、B、C分别为患儿、患儿父亲、患儿母亲
3 讨 论
3.1 SOTOS综合征患者的临床表现
超过90%的SOTOS综合征患者具有以下临床特征[3]:①特殊面容,如前额凸出、鼻梁低平、轻度斜视、高腭弓、眼距增宽、眼裂下斜、小下颌、尖下巴及额顶区毛发稀疏等;②学习障碍,如早期发育迟缓,轻度至重度智力障碍;③过度生长,如在青春期前,尤其1~6岁,头围或身高大于正常同龄儿第97百分位,或平均值的2个标准差;在青春期后,身高增长趋于正常,但大头畸形非常典型;④骨龄超前。其他临床表现还有脑室扩大、早期喂养困难、新生儿高胆红素血症、肌张力减退、新生儿低血糖、皮肤错构瘤、脊柱侧弯、关节过度松弛、扁平足、癫痫、便秘、肿瘤、淋巴水肿、先天性心脏病(动脉导管未闭、室间隔缺损及房间隔缺损等)及泌尿生殖系统异常(膀胱输尿管反流、隐睾、多囊肾)等[4-9]。成年SOTOS综合征患者有一个独特的面部特征,呈方形且突出的下颌。一般成年患者都很健康,仅少数患者会出现淋巴水肿、牙列不全、听力丧失、肢体挛缩和震颤等临床症状[10]。此外,SOTOS综合征患儿易合并孤独症、注意力缺陷与多动障碍、焦虑行为以及语言障碍等[11-15]。
本研究报道的该例5月龄的患儿,存在典型的SOTOS综合征临床表现,且颅脑MRI信号异常,可能存在神经系统异常。因家属拒绝骨龄检查,尚不能明确是否存在骨龄超前。该患儿经基因检测,发现5号染色体q35.3区出现NSD1基因区域错义突变c.5791 T>C,使该位点的半胱甘酸转变为精氨酸(p.Cys1931Arg),生物信息学软件预测其致病性很高,此突变点既往未有文献报道。结合基因检测结果,患儿符合SOTOS综合征诊断。
3.2 SOTOS综合征患者基因型与表型的关系
SOTOS综合征主要是由于NSD1基因单倍剂量不足所致,包括NSD1基因内突变(错义突变、无义突变、移码突变等)和5q35微缺失[1,16]。SOTOS综合征基因型在种族之间存在差异,于欧美国家中,NSD1基因突变型占80%~85%,5q35微缺失型占10%~15%;而在日本患者中,5q35微缺失型占50%以上[1]。本研究中的患儿NSD1基因存在未见报道的杂合突变(c.5791 T>C),临床表现与文献报道相似。中国内地共报道了17例SOTOS综合征患儿,有15例经基因检测确诊,其中NSD1基因突变型8例(4例错义突变,3例移码突变,1例无义突变),占47%(8/17);5q35微缺失型7例,占41%(7/17),均为散发病例,未见有家族性报道[17-25]。中国香港地区报道了36例SOTOS综合征,26例患儿NSD1基因异常,其中NSD1基因内突变型23例,占64%(23/36),5q35微缺失型只有3例,仅仅占8%(3/36)[26]。按照我国人口数量,报道的样本量过小,不能明确我国患者的基因型,说明我国对此类罕见病的临床认识不足。有研究报道5q35微缺失型与NSD1基因突变型之间存在表型差异,与基因突变型相比,5q35微缺失型患者更容易出现学习困难及心脏、肾脏异常等问题,而过度生长现象不明显[27]。在我国报道的17例患儿中,大部分存在特殊面容、过度生长及智力障碍等临床表现,几乎所有的患儿颅脑MRI显示脑室扩大、脑发育不良,部分患儿合并骨龄超前、低糖血症、腭裂、新生儿喂养困难、新生儿黄疸等。本研究报道的基因内突变型患儿临床表现与国内报道的相符,早期合并喂养困难及新生儿黄疸,无心脏及肾脏异常。在国内报道的17例患儿中,有4例患儿合并先天性心脏病或肾脏异常,皆为5q35微缺失型,与既往文献报道相符,可能微缺失型患儿更易合并心脏及肾脏异常。康路路等[17]报道了3例NSD1基因突变型患儿中,有1例患儿无特殊面容及过度生长。赵敏[20]报道了3例SOTOS综合征患儿,2例NSD1基因突变型无过度生长。孙碧君等[25]报道了2例微缺失型SOTOS综合征新生儿,1例出生时即表现为过度生长,另1例在随访至45个月时出现过度生长,与国外研究有差异,可能与我国样本量较小有关系。BOU-ASSI等[28]研究报道了1例以新生儿皮肤松弛症作为首发症状的SOTOS患儿。SIO等[29]报道了首例SOTOS综合征合并先天性巨结肠患儿。SOTOS综合征新生儿期的症状更加不典型,合并罕见临床症状时,可考虑存在基因异常。相关研究表明,高胰岛素血症是新生儿期SOTOS综合征患儿的特征性症状[30-31]。GRAND等[32]报道了7例合并高胰岛素血症的SOTOS综合征患儿。在高胰岛素血症或低血糖的背景下,即使无过度生长表现,也可以考虑此病[33],并应进行早期的基因诊断,以改善患儿预后。LACCETTA等[34]报道了1个家系的SOTOS综合征,该家族3代成员携带相同突变位点,先证者具有典型SOTOS综合征表现,母亲与外祖父仅身高大于正常同龄人第97百分位,无其他异常。韩国报道了1对同卵双生子患儿,2名患儿均为NSD1基因无义突变,先证者存在癫痫发作、脊柱侧凸、脑室扩大、注意力缺陷、多动障碍等异常的临床表现,而其胞弟并无上述表现,仅有SOTOS综合征的典型临床表现[35]。因此,突变位置与临床表型之间没有绝对的相关性,同一突变位点也可以表现出不同的临床特征。SOTOS综合征相关的临床特征,可能与基因型无关,具体机制尚不清楚,有待进一步研究。
3.3 SOTOS综合征的产前诊断及随访
如果产前超声显示胎儿生长过度、头颅畸形、羊水过多且合并其他异常,如泌尿系统异常、中枢神经系统异常、颈部透明带增厚和异常的母体血清筛查结果,应考虑此病,并应早期进行基因检测,有助于遗传诊断和咨询[36-37]。目前此病尚无特殊治疗方法,儿科定期随访很重要,以便早期处理并发症,提高患儿预后。
