Iron-regulation and Iron Chelation Therapy
2021-02-10
(Pharmacy and Pharmacology Department, University of Bath, BA2 7AY, UK)
Abstract: As an essential element, iron is able to assist enzymes to complete many biological activities in the body of human beings. According to Fenton chemistry, the generation of reactive oxygen species caused by iron increases the risks of cellular damage. It is therefore necessary to control the balance of the intracellular iron levels in our body. Moreover, iron is highly required in dividing cancer cells, suggesting that iron metabolism exerts significant impacts on tumour cell biology. However, the development of several useful clinical iron chelation agents such as desferrioxamine (DFO), thiosemicarbazone (5-HPCT and 3-AP) and deferiprone (DFP) has been limited in its widespread use in clinical practice because of low lipophilicity and high molecule weight. Nevertheless, some of the iron chelators from the hydroxypyridin-4-one (HPO) family notably, CP94 and CN128 show greater bioavailability and better physiochemistry properties, which provides a foundation of iron chelation therapy for skin cancer treatment.
Key words: iron; iron chelator; oxygen species; iron levels; skin cancer; tumour
Iron is an essential element that can assist the iron-related vital enzymes in the body. Iron-sulfur clusters complex Ⅰ and Ⅱ can be found in many enzymes. These complexes participate in crucial biological activities, including ATP generation and redox reactions[1].
Iron has been found to switch between two oxidative states (ferric- and ferrous) in our body by donating or accepting an important electron, which generates the production of reactive oxygen species (ROS). Importantly, the generation of ROS potentially increases the risk of cellular toxicity and damage. Therefore, it is necessary to keep the balance of intracellular iron levels via balancing iron uptake, usage and storage[2]. Similarly to the previous folate studies, iron is important in cancer development since it is highly required for an iron-containing enzyme, ribonucleotide reductase (RR), which plays a rate-limiting role in DNA synthesis[3]. However, a few investigations have been specifically designed and developed on iron chelators for the treatment of cancer.
Iron chelators are commonly used to treat iron-loaded diseases, particularly for β-thalassemia disease, but the development of iron chelators has been limited because of their low lipophilicity and high molecule weight like desferrioxamine (DFO)[4]. As for chelator design, the commonly used electron donor atoms are oxygen, nitrogen and sulfur, which are demonstrated to affect biological activity of agents[5]. Rapid glucuronidation leads to the placement of a non-chelating metabolite, and this is the main reason of lower efficacy of commercially available iron chelator deferiprone[6]. Another clinically useful iron chelator is CP94, which significantly enhances the efficiency of photodynamic therapy-aminolevulinic acid (PDT-ALA) combination therapy in basal cell carcinoma (BCC) clearance treatment[7]. Moreover, a novel iron chelator CN128 has been found to be a great potential in the treatment of clinical skin cancer with good oral bioavailability and tissue distribution[8].
In a word, iron chelation therapy is a potential cancer treatment. The aims of this review to discuss are: (ⅰ) the crucial role of intracellular iron-regulation; (ⅱ) iron metabolism in neoplastic cells; (ⅲ) the principles and potential uses of iron chelators agents as clinical chemotherapy agents for cancer treatment; and (ⅳ) the types of new members currently being considered for antitumour therapy.
1 General mechanism of iron regulation
1.1 Two oxidation states of iron
Iron is the foundation in sustaining our life. There are two oxidation states of iron that can be found in our body: the ferrous (Fe2+) and the ferric (Fe3+)[9]. By accepting or donating electrons, the ability of iron to switch between these two oxidation states is a key factor that enables it to perform a variety of biological functions[10].
Fe2++ H2O2→ Fe3+·OH + OH-,
(1)
Fe3++ reductant → Fe2+oxidized reductant.
(2)
According to Fenton chemistry, the generation of reactive oxygen species depends on the iron ability in redox cycle(Eq.(1)).If Fe3+reacts with a range of reductants, the iron (Ⅱ) can be produced through Eq.(2). Importantly, the mutation and cellular damage may result from the production of ROS and ·OH which leads to the reaction of lipids, proteins and DNA[10]. Thus, iron has been considered as a potentially toxic element.
1.2 Regulation of iron metabolism by iron-regulatory proteins
Once iron (ⅲ) is reduced to iron (ⅱ), it can be transported to the cells via the divalent metal iron transporter (DMT1) expressed on the tip of intestinal cells in the duodenum, which coordinates iron regulatory proteins 1 and 2 (IRPs)[11]. IRPs are straightforward to connect with iron uptake and efflux to control the intracellular iron level. These proteins play as the post-transcriptional regulators by regulating iron uptake via transferring receptor (TfR1) and iron sequestration via ferritin (Ft) to regulate intracellular iron homeostasis when the iron is sufficient or deficient (Fig.1)[11]. When iron is deficient, IRPs bind to the five IREs at the 3’ untranslated region (UTR) of TfR mRNA, which results in TfR mRNA stabilization. Meanwhile, IRPs bind to single IRE at the 5’ UTR of Ft mRNA, and the translation of Ft mRNA is inhibited, leading to decreased iron storage. However, when the iron is abundant, TfR mRNA binding activity of IRPs is lost, and the degradation of TfR mRNA can be found. The IRPs are found to bind to presented irons, which means that IRPs no longer bind to Ft mRNA and the translation of Ft mRNA is then increased. All in all, all these regulation mechanisms modulate intracellular iron levels directly or indirectly by affecting post-transcriptional proteins levels. A recently recognized transporter called heme carrier protein 1 (HCP1) is found in the apical membrane of the duodenal intestinal epithelium, which is related to dietary heme transportation[12]. However, there is no evidence demonstrating that this protein is a relevant physiological mechanism protein, although this protein seems to be transporting heme.
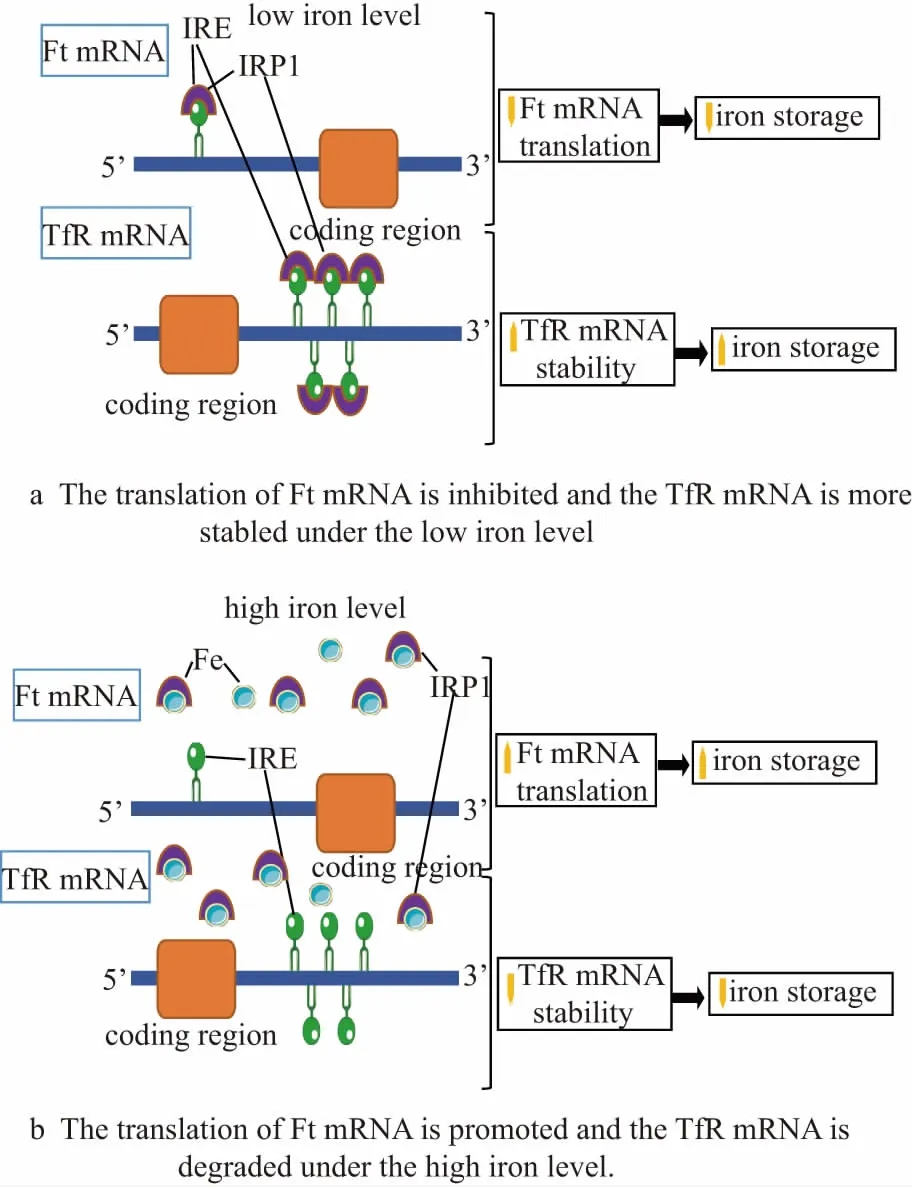
Figure 1 Schematic diagrams of iron metabolism regulation mechanism under the iron sufficient and deficient conditions
1.3 Iron transport and regulation of ferroprotein 1
Iron is transported into the intestinal cells and merges to form a pool of labile iron (LIP) composed of iron (ⅱ) and iron (ⅲ) in a redox equilibrium state. LIP performs a crucial role in iron export pathway associated with several important functions, including facilitating iron incorporated with proteins, being used as a chelator target, promoting free radical formation[12]. FPN1 located on the intestinal cells basolateral membrane is demonstrated to help iron to be exported into the circulation from the LIP[13]. The mechanisms of coordinating the release of iron and regulating FPN1 levels are related to IRP/IRE mechanism interaction and regulating protein levels by iron regulatory hormone and hepcidin, which is similar to IRP/IRE mechanism discussed above[14]. When the iron is deficient, the interaction between IRPs and IRE is removed at the 3’ end of the FPN1 mRNA, resulting in an increased FPN1 expression and the iron levels are therefore increased. The main reason for the reduced IRPs binding activity is that the 4Fe-4S cluster assembly in IRP1 or the IRP2 is reduced by iron in the proteasome. Additionally, the regulation of FPN1 expression is achieved by hepcidin that is mainly produced by hepatocytes[13]. After secreting from the liver, the FPN1 transporter internalization and its subsequent degradation on the basolateral surface of intestinal cells lead to iron accumulation in cells (Fig.2). High serum iron can induce hormone secretion, while low serum iron can inhibit the release of hepcidin. Therefore, when the serum iron is low or high, higher and lower FPN1 expression can be found, respectively[15].
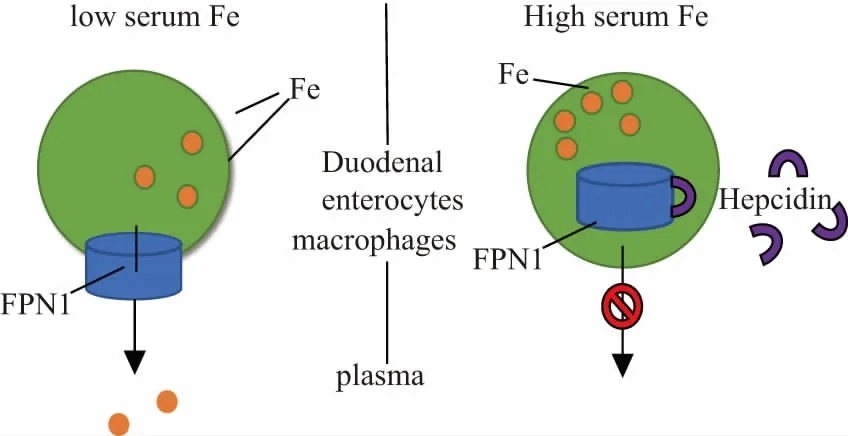
Figure 2 The influence of serum iron secretion level on hepcidin secretion and FPN1 final expression
2 Iron metabolism of cancer cells
2.1 Transferrin receptor 1 and 2 expression
Previous reviews have demonstrated several mechanistic differences in iron metabolism in cancer cells. One of the most important mechanisms is the up-regulation of TfR1 expression, which is a kind of glycoprotein, mainly expressed at the cell surface and can be found on all nucleated cells. Iron uptake is inhibited using monoclonal antibodies targeted to prevent Tf binding to the TfR1 proteins, leading to a significant antitumour activity in vitro[16]. The administration of phosphorothioate antisense TfR1 targeting the TfR1 mRNA sequence also caused the inhibition of tumour growth[16]. During incubation with iron chelating agents, under iron-depletion conditions, the up-regulation of TfR1 is found to increase iron uptake[17]. Together, iron chelating agents may bind to iron and target other molecular effectors preventing iron uptake, thereby inhibiting cell cycle progression leading to apoptotic cell death. TfR2 has also been found in various tumour models. In mice studies, compared with those only expressing TfR1 cells, an increased expression of TfR2 cells has been found in larger tumours[18—19]. Therefore, more interests in TfR2 targets are stimulated focusing on its role in regulating iron metabolism and tumour growth.
2.2 Ribonucleotide reductase and DNA synthesis
Ribonucleotide reductase (RR) showed a high requirement for iron. Compared with normal cells, an increased rate of DNA synthesis and cell proliferation is found in cancer cells, suggesting that the active RR plays an important role in neoplastic cells[20]. During DNA synthesis, RR which is a tetrameric enzyme participates in the rate-limiting step. RR is found to convert ribonucleotides to their 2’-deoxyribonucleotide counterparts to provide precursors for the DNA synthesis rate-limiting pathway[21]. There are two subunits consisted of RR (R1 and R2). R2 is an active site that contains a di-iron centre which is to stabilize the tyrosyl radical required to recuse ribonucleotides[22]. In the active site of RR, the ability of iron to cycle between iron (ⅱ) and iron (ⅲ) two oxidation states enables RR to act as an electron donor and acceptor, suggesting that the intracellular iron levels may determine the activity of RR directly.
Importantly, considering the function of RR in cellular proliferation, RR is confirmed to play an essential role. Specifically, an increased requirement of deoxyribonucleotides is found in the rapid development of tumour cells, particularly at the S-phase of cell cycle. Therefore, iron may also affect the expression of molecules which is involved in the cell cycles, controlling the cell cycle. Specifically, as for regulating the progression of cells from one phase to the next and regulating five phases G1, S, G2, M, and G0involved in the cell cycle, several regulated proteins like cyclins and tumour suppressor p53 provide potential antitumour targets to control cancer cell cycle progression[23—24]. Briefly, using an iron-chelating agent to consume iron will result in the differential expression of a series of cell cycle molecules. And then these changes will be observed with the cell cycle G1/S arrest and apoptosis[25]. Together, in order to contribute to anti-proliferative activity, the proper iron levels in the cell cycle progression are necessary, while the iron chelators are indicated that can target key iron regulatory molecules involved in the cell cycle. Additionally, Iron is vital for the process of angiogenesis and metastasis in cancer cells by coordinating with hypoxia-inducible factor-1 (HIF-1) system[26]. There are some adhesion molecules expressions on cell surfaces that have been demonstrated to be influenced by iron chelators, but the mechanism of how or whether iron-chelator agents affect the metastasis of cancer cells in vivo remains to be investigated.
3 Iron chelation in cancer therapy
3.1 Desferrioxamine (desferal®, DFO)
Desferrioxamine (DFO) (Fig.3a) is a natural hexadentate siderophore that is found in the streptomyces pilosus bacterium. As for iron-loading diseases like hemoglobinopathy and β-thalassemia, DFO is commonly used as the gold standard treatment since it is always bound to ferric iron with a higher affinity compared with other cations[5].This result is because DFO maintains three hydroxamic acid iron-binding moieties. As for DFO iron (ⅲ) complex, it not only shows great stability but also can form a 1∶1 stable complex (Tab. 1). This is the main reason that leads to the inhibition of accessing hydrogen peroxide or oxygen with iron centre directly so as to play its role[27].

Figure 3 Chemical structures of DFO and thiosemicarbazone series compound
Although the membrane permeability of DFO is poor, it can remove stored iron from ferritin which is the iron storage protein. In fact, in a range of cell types, DFO-treatment has been confirmed to maintain ability to regulate iron levels in vivo by inhibiting iron uptake and promoting iron release. However, the administration of DFO did not significantly affect haemoglobin levels when measuring mice studies, but DFO has been found an ability to reduce liver iron storage[28]. Additionally, as for clinical application is concerned, the DFO is limited due to its low bioavailability, and DFO shows a short plasma half-life (only about 12 minutes), and it needs strict administration when using DFO therapy through subcutaneous infusions[28]. Together, although DFO is a good iron chelation agent which provides a great choice to treat iron-loading diseases, its development for use as an antitumour drug is limited by some side effects such as poor potency, low membrane permeability and short plasma half-life.
3.2 5-Hydroxypyridine-2-carboxaldehyde thiosemicarbazone and 3-aminopyridine-2-carboxaldehyde thiosemicarbazone(5-HPCT and triapine®)
A clinical used iron chelator anti-tumour drug is thiosemicarbazones, but some mechanisms of its activity and actions are still unclear. Thiosemicarbazones is a tridentate ligand that possesses tridentate soft donor coordination systems N, N, S, which are essential for its anticancer activity[29]. The production of redox-active iron complexes formed by this ligand also plays a critical role since it could generate ROS pathways. Compared with DFO, some thiosemicarbazones exhibit cytotoxicity when they are forming iron complexes without significant anti-proliferative and cell loss activity.
A thiosemicarbazone-based structure 2-formy-
lpyridine thiosemicarbazone (Fig.3b) has been found in 1956 to treat leukemic. Another similar iron chelator agent 5-hydroxypyridine-2-carboxaldehyde thiosemicarbazone (HPCT) (Fig.3c) has been studied clinically to test its toxicity. The efficacy of this class of iron chelation was significantly observed since the characteristic dark-green urine was found after treating patients, which may result from the excretion of the iron (ⅱ) HPCT complex[30]. Additionally, three of five patients were shown a decrease in blast counts, but there was no anti-cancer activity or slow tumours development can be found in patients with solid tumours. However, some side effects have also occurred in HPCT treatment studies like mild myelosuppression activity, gastrointestinal toxicity and haemolysis[30].
One of the best characterized anti-tumour iron chelation thiosemicarbazones 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP), also called triapine®(Fig.3d), was initially designed as RR inhibitor since this may exhibit by ROS generation which is produced via the depletion of cellular iron pools or 3-AP iron complex. However, the real mechanism of this inhibition of tumour growth is still unknown. Compared with hydroxyurea, 3-AP has stronger proliferation and cell loss activity for leukaemia cells both in vitro and in vivo, especially for inhibited hydroxyurea-resistant cells[31]. This result indicated that 3-AP may display a different mechanism of antiproliferative and cell loss activity from hydroxyurea. Additionally, according to the results of Phase ⅱ clinical trial, the clinical use of 3-AP is limited by its cellular toxicity and low efficacy[3, 31].
4 Novel potential clinical iron chelation
4.1 Deferiprone and combined therapy
In order to learn more about novel iron chelator deferiprone (DFP) (Fig.4a) action in our body, the first clinical trials were measured on acute leukemia patients and thalassemia patients. The results showed that DFP increased urinary iron excretion, which also showed an increase in drug doses[6]. As for assessed the times of transfusions received for patients, an increasing iron load phenomenon was exhibited, and the studies found a poor correlation with serum ferritin levels[5]. Importantly, the clinical used DFP can be absorbed rapidly at the peak after ingestion for 45 minutes[6]. The speed of drug absorption may be slowed down by food, but the total amount of absorption would not be affected. However, the speed of glucuronidation will determine the amount of iron chelation, and with the duration of administration, there is no change of the speed of absorption, glucuronidation and clearance in the blood can be found[6]. In situations where deferoxamine treatment is contraindicated and insufficient, DFP-treatment is now allowed to treat iron-loaded diseases worldwide. It has been demonstrated that being treated initially with a lower dose can effectively reduce the risk of gastrointestinal side effects compared with being treated with a maximum dose[32]. Moreover, DFP can be used to regulate both liver and cardiac iron levels preventing their damage. A crucial role for iron chelation to prevent iron-induced cardiac damage is removing non-transferrin-bound iron from plasma, which can quickly be scavenged by deferiprone. Furthermore, the combined therapy of deferiprone and deferoxamine has been proved to be effective in solving the problem of cardiac iron overload and the impairment of left and right ventricular function[33]. Although deferoxamine has been suggested as a chelation agent for children and adults in recent years, the use of deferoxamine for iron chelation therapy is still limited by its side effects, such as agranulocytosis and gastrointestinal symptoms[33].
4.2 1, 2-Diethyl-3-hydroxypyridin-4-one hydrochloride (CP94)
There is a sophisticated method for ablating tissue called photodynamic therapy (PDT), which includes activating a photosensitizing drug via visible light and then producing activated oxygen in target cells, leading to their damage[34]. As for PDT in dermatology, topical active agents are preferred and make the photosensitizer 5-aminolevulinic acid (ALA) into a cream[7]. The ALA which can be found in all nucleated cells, is demonstrated as a precursor in the haem biosynthesis pathway. Moreover, the formation of the endogenous photosensitizer protoporphyrin IX (PpIX) results from the use of ALA, which usually binds to iron to form haem[7].
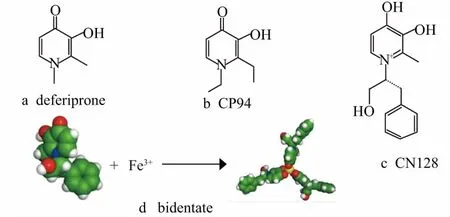
Figure 4 Chemical and crystal structures of DFP, CP94 and CN128
ALA-PDT treatment for skin cancer has received attention in recent years, especially for basal cell carcinoma (BCC)[7]. Briefly, since the size, site and multiple lesion presentation are found with BCC, this skin cancer is currently difficult to be treated. ALA-PDT may be a new window for the therapy of skin cancer, notably BCC. if the disease is superficial, the PDT treatment would show good results[7]. In order to improve the efficiency of ALA-PDT treatment, a novel iron chelator 1, 2-diethyl-3-hydroxypyridin-4-one hydrochloride, CP94 (Fig.4b) is considered to be used as the potential enhancer, suggesting that the hydroxypyridinones family is a new series with lower molecular weight and greater lipophilicity (Tab.1).

Table 1 Comparison of desferrioxamine(DFO), deferiprone (DFP), CN128 and CP94
The studies showed that the combination of CP94 and ALA-PDT therapy is safe without dose-limiting adverse events. Increasing the concentration of CP94 when added to ALA-PDT therapy decreased the pain endured by the patients during the light therapy[34]. Importantly, after CP94-treatment with only one cycle PDT, a complete clearance of nodular BCC was exhibited without prior lesion preparation, suggesting that CP94 provides a safer and simple pharmacological modification to ALA-PDT treatment[7, 34].
4.3 CN128
The identification of a novel iron chelator CN128 is achieved by using a novel strategy undertaking glucuronidation by a combination with an alternative sacrificial site. The space near the 3-hydroxypyridon-4-one nucleus is then explored guided by the LogP values of CN128[8]. CN128 has been demonstrated as a bidentate iron chelator (Fig.4c) with a similar iron (ⅲ) ability and transition metal selectivity compared with DFP (Tab.1). Importantly, CN128 has been demonstrated to maintain a great lipophilic distribution coefficient, a good tissue distribution, and satisfactory oral pharmacokinetic profiles, which showed a better potential in cancer treatment. Moreover, the 3-hydroxyl glucuronidation regulated effect which may relate to the efficacy of iron removing is confirmed to reduce with CN128. When oral dosing CN128 at 150 μmol/kg, the result showed that the iron removal efficacy was 3.8-fold greater than the same oral administration of DFP[8]. A significant side effect for DFP is the decreased white cell count, but this is not shown in the CN128 treatment, which not only has no change in the white cell population but also could increase its quantity after oral dosing for 2 months[8]. Together, these results indicate that CN128 is currently investigated as one of the most promising iron-chelation agent candidate for cancer treatment.
5 Conclusions
Iron serves as a critical element in many biological activities as it can affect the function of iron-containing enzymes. In this review, iron is demonstrated to maintain an ability to cycle transfer between iron (ⅱ) and iron (ⅲ) oxidations states by donating a vital electron via Fenton chemistry to sustain our life. This is one of the main reasons that potentially cause cell damage. Intracellular iron levels are regulated by IRPs and IRE mechanism acted as the post-transcriptional regulators to regulate iron uptake via transferrin receptor (TfR1) and iron sequestration via ferritin (Ft). The IRP-IRE mechanism also regulates FPN1 expression by iron regulatory hormone and hepcidin to achieve FPN1 internalization under low iron or high iron conditions. Importantly, the lack of iron may reduce DNA synthesis by affecting the rate-limiting enzyme RR in cancer cells. The TfR1 expression is also increasing when tumour growth, while TfR2 expression can be found in larger tumours. Further investigation about the iron regulation in DNA synthesis has confirmed that iron does not only have an influence on tumour cell angiogenesis and metastasis but also affects many molecules expression in cell cycles like CKIs, p53, CDK, etc which are involved in cancer cell biology.
Iron chelation therapy is demonstrated as one of the most potent strategies in cancer therapy. DFO is commonly used as the gold standard treatment for iron overload disease as it binds to ferric iron with higher affinity, but the membrane permeability of DFO is poor. As for the thiosemicarbazones class, iron-chelation agents HPCT and 3-AP which are initially designed as RR inhibitors showed a greater antiproliferative and cell loss activity. Moreover, DFP prevents liver and cardiac damage by regulating iron levels, but some side effects such as agranulocytosis and gastrointestinal symptoms may limit its development. CP94 and CN128 from the hydroxyprodline-4-one family are demonstrated to have the potential to be developed as iron-chelation agents as they exhibit greater efficacy, better oral pharmacokinetic profiles, lower toxicity and satisfactory distribution in our body. Together, iron chelation therapy provides a novel strategy for cancer treatment. However, the development of recently clinically used iron chelators has been limited because of their poor physicochemical properties and side effects. Several novel iron chelators have been identified and undertaken investigation as the potential candidates to treat cancer.
