五种治疗阿尔茨海默病药物分子结构与性质的密度泛函计算分析
2020-12-28严茜茜黄罗仪王朝杰向铮
严茜茜,黄罗仪,王朝杰,向铮
(1.温州医科大学 药学院,浙江 温州 325035;2.温州市第七人民医院 药剂科,浙江 温州 325000)
阿尔茨海默病(Alzheimer’s disease,AD)是一种起病潜隐,病症呈进程性渐进的神经退行性疾病,具有记忆衰退,认知恶化,执行能力丧失以及行为改变等特性。在高龄人群中易生成痴呆,占全部痴呆类型的近60%[1],严重降低当代老年人生活质量。AD病理复杂并伴多种代谢途径共同参与[2],其发病机制假说众多,主要有胆碱能神经元损伤机制[3],β-淀粉样蛋白级联机制[4]、τ蛋白过度磷酸化机制。2020年4月,美国南加州大学凯克医学院的研究团队又提出了新的血脑屏障破坏机制[5]。各机制之间相互联系,互为影响,错综复杂,尚无统一认识。目前临床用药品种少,主要有美金刚、多奈哌齐、石杉碱甲、卡巴拉汀、加兰他敏五种药物,且疗效有限,对治疗AD新药的研制有迫切需求。结构决定性质,对于已上市药物的结构研究可以更好地帮助我们了解药物的本质。理论方法是衔接分子结构与实验结果的桥梁,理论计算帮助我们从分子水平理解宏观性质。这五种抗AD药物在油性状态下的结构和性质,系统的整体和局部反应性描述未见报道,在模拟大脑环境下的计算研究将更助于本课题组从分子水平上理解抗AD药物的作用机制。
1 材料和方法
采用密度泛函理论(density functional theory,DFT)中杂化泛函B3LYP[6]方法,在TZVP基组水平上对五种药物进行几何结构优化和电子结构计算,在此基础上获得分子表面静电势(molecular electrostatic potential,MEP)、紫外-可见光谱(UV-Vis)和概念密度泛函系列指数。考虑到药物作用大脑环境,采用环己烷为溶剂进行模拟,运用导体极化连续模型(conductor-like polarizable continuum model,CPCM)进行模拟计算,计算工作通过G16[7]程序包完成,图像借助Gaussian View 6.0和Multiwfn3.6[8]软件进行分析和绘制。
2 结果
2.1 几何结构分析 将五种药物在B3LYP/TZVP水平下优化,得到最稳定构象及部分键长见图1。五种药物均具有环状疏水结构,如苄基、吡啶、苯环等,这与抗AD药物需要通过血脑屏障,具有一定的亲油性相吻合,可见抗AD药物有一定的结构共性,而分子本身结构的差异,又导致了其作用机制的不同。观察五种药物分子极性键的情况,以美金刚C4-N13为基准,当C原子上连有苯环时,C-N键长稍有缩短,加兰他敏C14-N15与多奈哌齐C17-N14键长均为1.459Å。当C原子双键连接O原子时,C-N键长缩短更加明显,石杉碱甲C12-N13为1.400Å,而卡巴拉汀C4-N3为1.354Å,相较之下键长更短,键能更高。由于N原子上的孤对电子与苯环的π电子形成了共轭效应,使得两个原子结合得更牢固,这种类似于肽键的结构印证了在N3-C4-O16之间存在共轭。朱维良等[9]等运用量子化学计算石杉碱甲单分子状态的键长数据显示C12-O14为1.217Å,C2-N18为1.538Å,N13-H29为0.997Å,而形成石杉碱甲-乙酰胆碱酯酶(acetylcholine esterase,AChE)复合物后C12-O14为1.256Å,C2-N18为1.477Å,N13-H29为1.030Å。我们在B3LYP/TZVP水平计算得到的数据与石杉碱甲单分子状态进行比较,最大键长差为0.066Å,与形成石杉碱甲-AChE复合物后比较,最大键长差为0.024Å。计算数据与形成复合物后的数据更相近。陈卫民等[10]通过单晶X-射线测定了卡巴拉汀晶体结构,测得N13-C18为1.490Å,N13-C12为1.525Å,C6-O5为1.405Å,C4-O5为1.360Å,C4-O16为1.203Å,与理论计算值相比,最大键长差为0.049Å。RAVIKUMAR等[11]测得多奈哌齐晶体结构中C1-O25为1.364Å,C2-O26为1.354Å,C9-O24为1.220Å,C8-C10为1.519Å,C7-C8为1.537Å,C10-C11为1.538Å,C17-C18为1.500Å,C17-N14为1.504Å,与理论计算值相比,最大键长差为0.045Å。理论数据和实验晶体结构数据相近。
2.2 MEP分析 MEP是量子化学用于研究分子反应性、分子与受体相互作用及其他现象的重要工具。通过MEP能直观感受到电子云分布状态,预测分子亲核亲电反应活性位点。五种药物在B3LYP/TZVP水平下的MEP见图2,美金刚整体含一处负的静电势,分布在氮原子周围,最小极值点亦在N13附近,其他部分呈正的静电势。加兰他敏整体有3处含负的静电势,主要分布在氧原子和氮原子周围。最小极值点在醚氧键和羟基附近,最大极值点在羟基附近。石杉碱甲整体有两处含有负的静电势,分别在氧原子和氮原子上,最大极值点在N13附近而最小极值点在O14附近。多奈哌齐整体有三处含有负的静电势,为茚酮部分、苄基部分和甲氧基部分。最小极值点在O24附近,最大极值点在C3附近。卡巴拉汀整体有两处含有负的静电势,分别在氮原子及氨基甲酸酯部分,最大极值点在N3附近,最小极值点在O16附近。拥有不同电性部位导致了五种药物分子理化性质的差异。

图1 五种抗AD药物在B3LYP/TZVP水平下的几何结构及部分键长数据(Å)
有研究表明加兰他敏的羟基能够以氢键的形式与酶发生作用[12],多奈哌齐吡啶环中的N质子化后可以在AChE的通道部分产生阳离子-π作用,同时与水产生氢键。石杉碱甲羰基氧与AChE催化活性位点(catalytic active site,CAS)形成氢键,氨基上的氮原子在AChE的通道部分形成氢键。氢键本质是强极性键上的氢原子与带有负电荷的原子之间的静电作用力,对于生物高分子反应有着重要的意义。而对于富含芳香环氨基酸的AChE来说,这五种药物也含有丰富的芳香环,因此发生在芳环之间的π-π堆积作用,与氢键一样是重要的非共价键形式。研究表明加兰他敏环己烯上的双键与AChE的CAS位点形成π-π叠加[13],多奈哌齐苄基部分可与AChE的CAS位点产生π-π堆积,茚酮部分与AChE外周阴离子位点(peripheral anionic site,PAS)形成π-π堆积[14]。结合五种药物的MEP不难发现电性差异决定了分子间作用力、作用位点的不同,电性极值点往往对应药物与酶作用的位点,这对改变蛋白质构象具有关键性的作用,是反应核心。石杉碱甲的合成研究也验证了这一点,天然提取的石杉碱甲构型为左旋的单一对映体,唐希灿等[15]将其对映异构体、外消旋体对AChE的抑制活性与之进行比较后认为右旋异构体活性和选择性都大为降低是由于构型不匹配,环外亚乙基甲基的氢无法与酶形成有效氢键。

图2 五种抗AD药物在B3LYP/TZVP水平下的MEP图
2.3 前线分子轨道分析 前线分子轨道(frontier molecular orbital,FMO)理论将分子周围排布的电子云根据能量细分为不同能级的分子轨道,指出其中的最高占据轨道(highest occupied molecular orbital,HOMO)简称H,最低空轨道(lowest unoccupied molecular orbital,LUMO)简称L,是决定一个体系发生反应的关键。△ε(L-H)表示HOMO与LUMO的能差,反应了分子形成过渡态所需的能垒大小,差值越小,则能垒越小,越容易发生反应,化合物活性也就越高。
五种药物的HOMO、LUMO及能差见表1,由表1可知,四种抗AChE药物之间略有差异,但活性明显高于N-甲基-D-天冬氨酸受体(N-methyl-D-aspartic acid receptor,NMDAR)拮抗剂美金刚。根据FMO,五种药物可能的反应性顺序为多奈哌齐>石杉碱甲>卡巴拉汀>加兰他敏>美金刚。

表1 五种抗AD药物分子的最高占据轨道和最低空轨道能及其能差
2.4 紫外-可见吸收光谱分析 五种药物UV-Vis数据见表2,美金刚的UV-Vis存在两个吸收峰,141.0 nm 处吸收强度大,跃迁类型属于σ→σ*跃迁。美金刚化学结构独特,分子结构中无共轭双键,一般紫外-可见分光光度计只能提供190~850 nm范围的单色光,因此在紫外区实验无法观测到,但是我们的理论计算能够提供UV-Vis数据。其余四种抗AChE药物的最大吸收峰落在179~208 nm之间,跃迁形式均为π→π*跃迁。李霞等[16]测得甲醇溶液中加兰他敏在289nm处有最大吸收。王晓燕[17]对氢溴酸加兰他敏标准品进行紫外扫描结果显示,在322、254及289 nm处有较强吸收。LIU等[18]测得乙醇溶液中石杉碱甲在231和313 nm处有最大吸收。胡征[19]测得多奈哌齐在甲醇溶液中206.6 nm处有最大吸收,强度为0.921,在0.1 mol/L的HCl溶液中207.4 nm处有最大吸收,强度为1.212。王晗宁[20]测得溶剂为乙酸乙酯时,多奈哌齐在264和315 nm处分别有吸收,其中264 nm处有最强吸收峰。郭淼[21]测得甲醇溶液中卡巴拉汀最大吸收波长210 nm,且在262 nm处有一个特征吸收峰。比较可知随溶液极性增加化合物吸收强度增大,整体发生红移现象。

表2 五种药物紫外吸收光谱数据
2.5 概念密度泛函分析 概念密度泛函理论适用于多电子体系结构的量子力学分析,是DFT的重要的分支,它把与化学相关的概念和原理从密度泛函中抽取出来,通过理论推导获得计算参数,赋予了从前已知意义但是相对模糊的化学概念一个相对精确的描述。提出了全局反应性指数,包括:电离势(IP)、电子亲和势(EA)、化学势(μ)、电负性(χ)、整体硬度(η)、整体柔软度(S)和整体亲电指数(ω)。
五种药物的全局反应性指数见表3。μ和η能体现分子的稳定状态,五种药物的η相差不大且皆为正值,表明对微干扰下化学体系电子云畸变的抵抗能力较强。多奈哌齐μ最低而加兰他敏μ最高,表明多奈哌齐更稳定。多奈哌齐的χ和ω较大,说明其得电子能力强。S反映了分子周围电子云分布情况,能体现化合物的反应活性,由S判断药物活性顺序为多奈哌齐>石杉碱甲>加兰他敏>卡巴拉汀>美金刚。郑清川等[22]运用人类乙酰胆碱酯酶与抑制剂小分子对接后发现与AChE相互作用的强弱顺序为多奈哌齐>石杉碱甲>卡巴拉汀,与我们由S判断的活性顺序相一致。在多奈哌齐、卡巴拉汀和加兰他敏治疗AD的安全性和耐受性比较研究中发现,多奈哌齐相较于其他两种药物胃肠道不良反应发生率更低,三种药物中枢神经系统和心血管系统不良反应均较少发生,临床应用多奈哌齐的依从性更好[23]。
在概念密度泛函中引入福井函数,以衡量当总电子数或外部化学势发生变化时的电子密度差异。为进一步认识药物分子活性情况对五种药物进行了福井函数计算。结果发现,美金刚的亲核部位具有偏向性,主要在N原子上连接氢原子的一侧以及底部下方的位置,亲电部位则不具偏向性,均匀分布在整个分子表面。加兰他敏的亲核部位同样具有偏向性,主要分布于右下侧苯环上,亲电部位则偏向于右侧苯环以及上方N原子部位。石杉碱甲的亲核部位偏向于左边的吡啶环上,亲电部位则均匀分布于整体分子。多奈哌齐的亲核部位主要位于左侧茚酮结构上,亲电部位主要在右边吡啶环一侧。卡巴拉汀亲核部位主要在中间苯环上,亲电部位主要在右边氨基甲酸酯结构,见图3。
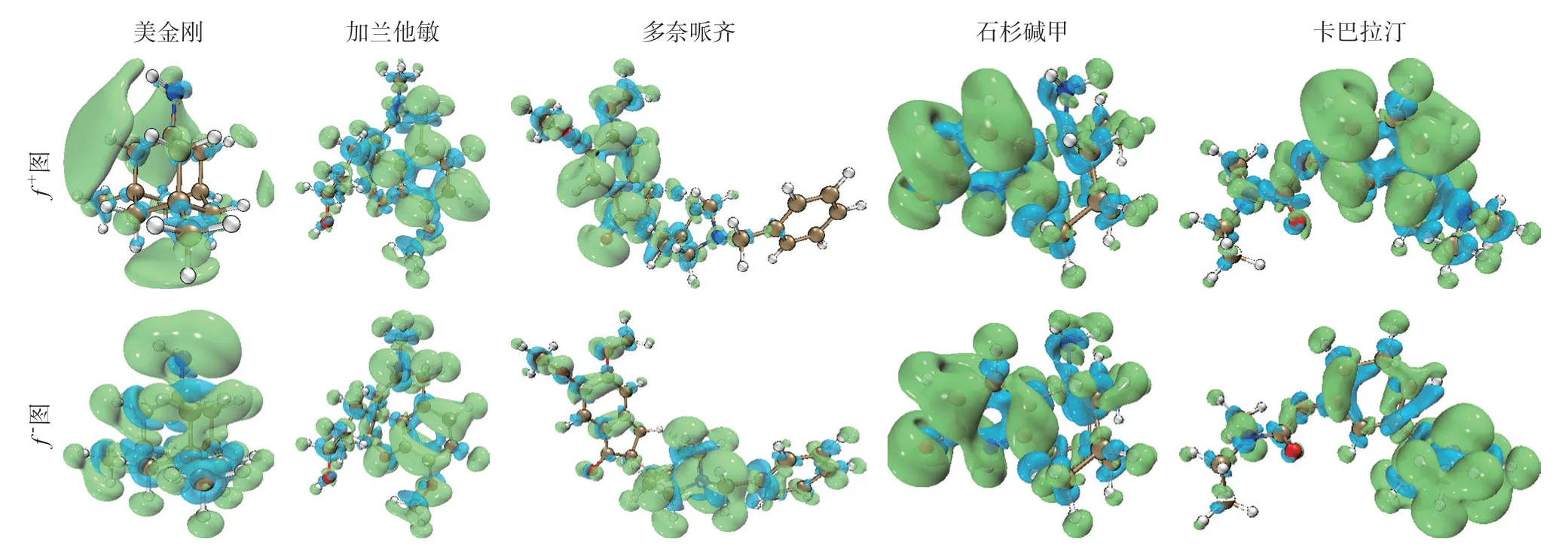
图3 五种抗AD药物的福井函数图
五种药物分子亲电位置的差异与他们结构中是否带有富余的电子密切相关。相对亲电指数最大值分别为美金刚H14位4.730,加兰他敏C5位6.391,石杉碱甲C10位2.923,多奈哌齐C9位23.071,卡巴拉汀C8位25.006。结合相对亲电指数来看,多奈哌齐和卡巴拉汀亲电能力强,美金刚、加兰他敏和石杉碱甲亲电能力较弱。亲核位置的差异不仅与分子所带电荷量相关,而且与分子结构的空间位置阻力也有着密切的联系,相对亲核指数最大值分别为美金刚N13位12.514,加兰他敏N15位203.804,石杉碱甲C8位2.602,多奈哌齐N14位152.297,卡巴拉汀N13位86.031。结合相对亲核指数来看,加兰他敏、多奈哌齐、卡巴拉汀的亲核能力强,美金刚和石杉碱甲亲核能力较弱。
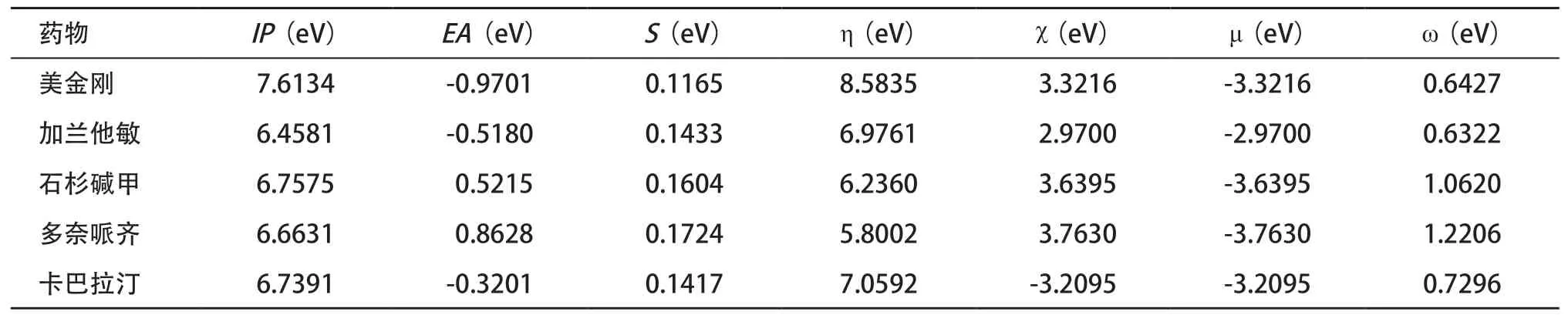
表3 五种抗AD药物在B3LYP/TZVP水平下的全局反应性指数
3 讨论
本研究在B3LYP/TZVP水平对美金刚、加兰他敏、石杉碱甲、多奈哌齐和卡巴拉汀的分子结构、电子结构、UV-Vis以及概念密度泛函进行分析,结构分析比较出形成电子共轭效应使得极性键键长缩短。同时显示计算数值与实验数据吻合较好,表明借助DFT在B3LYP/TZVP水平可较好的研究抗AD药物。电子结构分析显示电性差异决定了分子间作用位点不同,特别是电性极值点,往往对应着与蛋白质形成非共价键的位置,是药物作用的关键,这对今后抗AD药物作用位点研究可以提供一定的指导作用。前线轨道分析显示五种药物中四种抗AChE药物活性略有差异但明显高于NMDAR拮抗剂,UV-Vis分析结果显示溶剂效应使分子的吸收波长发生红移且吸收强度增强。全局反应性指数表明多奈哌齐为五种抗AD药物中软度最大、电负性最大、亲电指数最大、化学势最低的药物,药理作用最强。由福井函数结合相对亲电指数与相对亲核指数得出多奈哌齐和卡巴拉汀亲电、亲核能力都很强,加兰他敏亲核能力强,美金刚和石杉碱甲亲电、亲核能力均比较弱。本研究结果为深入探索抗AD药物作用机制以及药物活性比较提供一定的计算支持。
