哺乳动物细胞中糖链结构的改造
2020-05-11金则成喜多岛敏彦藤田盛久高晓冬
金则成, 喜多岛敏彦, 藤田盛久, 高晓冬
(江南大学 生物工程学院,江苏 无锡214122)
生物药物分子是一类由生物体包括细菌、酵母、昆虫、植物和哺乳动物细胞生产的药物。 药物蛋白质如单克隆抗体具有高特异性和与靶分子的高亲和性, 同时与传统药物相比具有更低的副作用。目前,重组药物蛋白质已经在治疗疑难杂症如癌症和自身免疫疾病方面变得越来越重要[1-3]。 目前,大部分药物蛋白质主要由哺乳动物细胞合成,这主要是由于哺乳动物细胞生产蛋白质可以提升正确折叠蛋白质的产量和促进翻译后蛋白质的折叠[4-5]。 由于大多数分泌蛋白质具有糖链,为了维持蛋白质的稳定性和功能以及抵御免疫反应,适当的糖链是非常必要的。
使用哺乳动物细胞生产生物药物分子依然有几个缺点,包括高成本和糖链的不均一性。 为了确保用作为生物药物分子的蛋白质质量和稳定性,糖链的不均一性是一个必须要解决的问题。 举例来说,红细胞生成素和粒细胞集落刺激因子(G-CSF)之类的细胞因子在体外必须具有含唾液酸的复合型糖链才具有活性[6-9]。 同时,对蛋白质的糖链结构进行改造可以提高蛋白质的功能活性。 举例来说,具有移除岩藻糖结构的N-糖链的抗体可以大幅提高抗体依赖性的细胞毒性(ADCC)作用[10-11]。 现在,为了生产抗体,不进行岩藻糖修饰的细胞已经被频繁地使用。 可以生产均一的糖蛋白的哺乳动物细胞株的构建是目前生物医药品生产领域中最迫切的需求。
图1 展示了糖链在哺乳动物细胞中的合成途径,糖蛋白首先在内质网中合成,随后被转运到高尔基体中,蛋白质上的糖链在α-1,2 甘露糖苷酶的催化下转变成Man5-GlcNAc2 型结构的糖链。 目前获得Man5-GlcNAc2 型结构的糖链主要有两种方法:一种是利用N-乙酰葡糖胺转移酶I(GnT-I)编码基因MAGT1 的突变或敲除细胞生成, 然而这种方法的缺点是该细胞株不能产生Man9-GlcNAc2或Man8-GlcNAc2 型结构的糖链;另外一种方法就是通过化学试剂几夫碱(kifunensine)或脱氧野尻霉素(deoxynojirimycin)抑制细胞α-1,2 甘露糖苷酶的活性,缺点是如果想要获得稳定均一的糖型,需要用化学试剂持续处理细胞。
溶酶体中含有多种水解酶,这些水解酶大部分是带有糖链的糖蛋白,它们可以将蛋白质、粘多糖、糖脂等物质消化为小分子,提供细胞再次回收利用[13]。这些水解酶在内质网中合成,在高尔基体内进行糖链的修饰,然后通过特定的M6PR 受体的识别被转运至溶酶体中。 高尔基体中糖链的修饰往往是通过在核心糖链的Man 的6 位上添加UDP-N-乙酰葡萄糖胺(UDP-GlcNAc)的N-乙酰葡萄糖胺-1-磷酸酯部分(GlcNAc-1-P),产生Man-6-P-1-GlcNAc,然后将GlcNAc 的部分除去, 形成具有酸性糖链的糖蛋白,然后通过特定的M6PR 受体的识别被转运至溶酶体中[14-16]。

图1 哺乳动物细胞N-糖链合成途径Fig.1 Pathway of N-glycan synthesize in mammalian cells
当产生的水解酶因为代谢通路异常无法正常运送至溶酶体或者是因控制该溶酶体酶的基因发生突变,该酶反应链中的中间产物不能正常降解而在溶酶体中贮积,从而引起细胞组织器官功能的障碍,导致溶酶体贮积症的发生[17-18]。 例如,法布莱病的病人由于缺少α 半乳糖苷酶,使得糖脂特别是一种中间产物globortriaosylceramide(Gb3)无法被分解而蓄积在细胞的溶酶体中, 从而威胁着他们的生命。 目前对于溶酶体贮积症主要的疗法有:酶替代疗法、化学疗法、基因水平上的基因修饰疗法等,而其中最为经典的方法即为酶替代疗法。 由于细胞膜表面存在M6PR,M6PR 可以识别药物蛋白质上的糖链结构并将蛋白质带到溶酶体, 从而能够通过M6PR 用正常的水解酶替代自身损伤的水解酶,使溶酶体贮积症得到改善[19-20]。
因糖链的不均一性而导致的糖蛋白的不均一性会对糖蛋白的生产及其应用产生不良影响。 由于M6PR 对于糖蛋白具有专一识别性, 当糖蛋白中存在部分糖链结构并非高甘露糖型或者说不存在6位被磷酸化的糖链时,会导致M6PR 对于药物的吸收效率的降低,导致治疗效率不高[21-22]。 另外,当糖链存在不均一的情况时,因为糖链的结构还可能导致糖蛋白被机体识别为外来的抗原物质,从而引起免疫反应。 出于药物分子的安全性的考虑,需要尽量确保糖链的均一性。
本研究可提供一种可用于生产均一性药物糖蛋白的细胞株, 即敲除MAN1A1 和MAN1A2 的双敲除细胞株Double-KO 细胞株。该细胞株中高甘露糖型N-糖链所占比例增加,同时由于复合型N-糖链的下降也可以证明该细胞株内糖链的表现形式更均一。
1 材料与方法
1.1 细胞培养
人体胚胎肾细胞HEK293 使用10%胎牛血清(FCS) 的DMEM 培养基 (Gibco,Life technologies,USA)培养,培养条件为37 ℃、5%。
1.2 高尔基体α-甘露糖苷酶的基因敲除质粒的构建
利 用 CRISPR -Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats)技术[23]敲除基因往往需要设计一个20 bp 长度的序列片段, 且该序列片段后要有一个PAM 位点(NGG/NAG)。 在本实验中,需要敲除的两个基因MAN1A1/MAN1A2 的基因序列从NCBI 上下载得到。 关于guide-RNA 的设计, 在Michael Boutros lab's Target Finder(http://www.e-crisp.org/E-CRISP/designcrispr.html) 上找到敲除基因所需的guide-RNA 的DNA 序列。 通过网站设计可以获得MAN1A1 的两条靶序列及各自所使用的引物序列为:
MAN1A1KO1:AAAACCACGAGCGGGCTCTCAGG PrimerKO1F:caccAAAACCACGAGCGGGCTCTC PrimerKO1R:aaacGAGAGCCCGCTCGTGGTTTT MAN1A1KO2:CCACCTTCTTCTTCTCCAGTAGG primerKO2F:caccCCACCTTCTTCTTCTCCAGT
primerKO2R:aaacACTGGAGAAGAAGAAGGTGG MAN1A2 的两条靶序列及各自所使用的引物序列为:
MAN1A2KO1:CCTTTACCGGCATCTACATGTGG rimerKO1F:caccCCTTTACCGGCATCTACATG rimerKO1R:aaacCATGTAGATGCCGGTAAAGG AN1A2KO2:CATGGATCAGGAAGACTCCGGGG rimerKO2F:caccCATGGATCAGGAAGACTCCG rimerKO2R:aaacCGGAGTCTTCCTGATCCATG。如图2 所示,将含有CRISPR-Cas9 系统的质粒pX330-EGFP 使用Bbs1(NEB:R0539S)切开,并使用Mighty Mix 将设计好的靶序列与质粒上guide-RNA 的DNA 序列序列相连接, 构建为含有MAN1A1/MAN1A2 靶点的质粒, 并将它们命名为:pX330 -EGFP -MAN1A1KO1/pX330 -EGFP -MAN1A1KO2、pX330-EGFP-MAN1A2KO1/ pX330-EGFP-MAN1A2KO2。
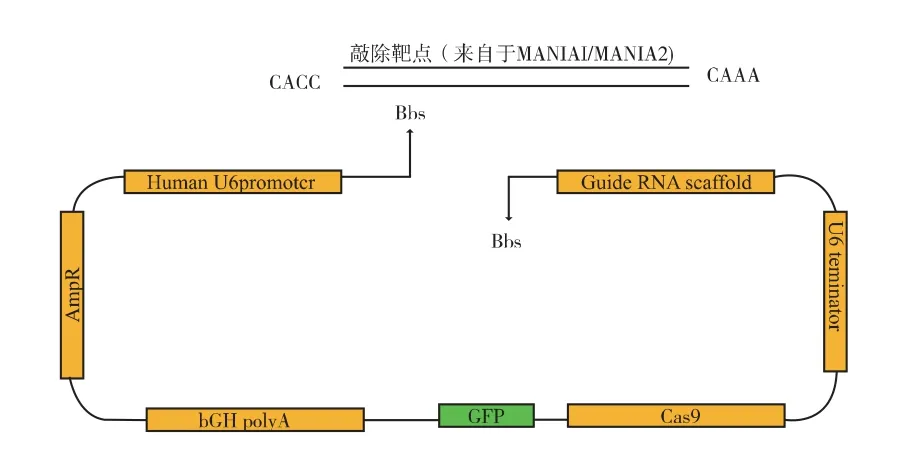
图2 CRISPR/Cas9 敲除质粒的构建Fig. 2 Construction of CRISPR/Cas9 KO plasmid
1.3 转染
将野生型细胞HEK293 使用10%FCS 的培养基培养过夜, 待其生长到约90%~95%时进行转染。转染试剂使用PEI-MAX[24](2 mg/mL pH 7.5),在转染前先需要将PEI-MAX 与OPTI(life technologies:31985-070)混合均匀,其比例为1 μL PEI-MAX∶50 μL OPTI 培养基。将敲除所需质粒和携带抗性基因的质粒pME-puro 与OPTI 培养基混合均匀,质粒添加量的比例为4 μg DNA∶5 μL PEI-MAX。将PEIMAX 溶液和含质粒溶液混匀,常温放置25 min,使质粒与PEI-MAX 结合。 之后将混合的溶液加入野生型细胞株的培养基中。 12 h 更换新鲜培养基,待生长恢复后(约24 h),更换为质量浓度1 μg/mL 的嘌呤霉素的培养基进行筛选。
1.4 单克隆细胞的获得与验证方法
筛选得到的细胞含有抗性质粒和敲除质粒,使用限制性稀释使单个细胞生长于96 孔板中, 获得单克隆细胞。 当细胞数量增加后将单克隆细胞转移至12 孔板培养。 当其生长为100%状态时,移去培养基,使用PBS 冲洗一次,加入胰蛋白酶(Sangon,CN)消化细胞,加入含10%FCS 的DMEM 培养基收获细胞。 所得细胞液于3 000 r/min 离心2 min,并再次使 用 磷 酸 缓 冲 液 (phosphate buffered saline,PBS,Sangon,CN) 冲洗得到沉淀物。 在沉淀物中加入50 mmol/L NaOH 并于金属浴中95 ℃反应20 min,反应结束后加入1 mol/L 的Tris (pH 7.5) 于15 000 r/min离心3 min,取上清液待用。 验证方法使用测序验证的方式,将提取的基因组使用验证设计的验证引物进行PCR 验证反应, 反应后的PCR 产物寄送华大基因测序公司进行测序。 设计的验证引物为:
check MAN1A1F:TTCCTGCCAGACTCCTCCAAG;check MAN1A1R:CCACTCACCTCTTTGATCTTTGC C;check MAN1A2F:CTTAGTGCCTTCATCACTCTGT GT;check MAN1A2R:GACAGATTCGATCCAATCAC CGT。
1.5 流式细胞分析
细胞被胰蛋白酶消化处理后收集细胞液,用磷酸缓冲液洗涤1~2 次。 取5×105个细胞重悬于流式细胞分析液FACS(PBS,1% BSA 和0.1% )中,加入带有荧光标记的凝集素, 如PHA-L4-FITC 和ConA-FITC(J-oil mills,Tokyo,Japan),使其终浓度为1%,反应约15 min。 使用3 000 r/min 离心3 min后弃去上清液, 使用FACS 洗涤2 次后再次离心移去上清液并重悬于FACS 溶液中。 使用BD Accuri C6(BD,USA)流失细胞分析仪进行分析。
1.6 细胞株中LAMP2 蛋白糖链变化的免疫印迹法验证
将细胞通过细胞裂解液裂解, 置于冰上孵育30 min。将细胞裂解提取液中蛋白质变性,使用糖类内切酶H(Endo-H,New England BioLabs)或PNGase F于37 ℃处理3 h。 处理后的蛋白质样品在SDS-PAGE中上样, 然后转膜到PVDF 膜上。 使用鼠单克隆anti-LAMP2 抗体用于检测细胞裂解提取液中的LAMP2 蛋白质[25]。
1.7 细胞生长曲线的测定
将细胞消化后使用细胞计数器计数。 将所得的细胞稀释至2 000 个/dL 的细胞浓度,将该浓度的细胞分装至96 孔板中,每孔体积为100 uL,将细胞放置于37 ℃、5%的条件下培养。 6 h 后在每孔内加入CCK-kit(Dojingo,Japan)后培养箱内培养1 h 后,在450 nm 纳米波长下测定吸光度变化[26],该结果即为0 h 数据, 之后每隔24 小时重复使用CCK-kit 测定,记录下0、24、48、72、96、120、144 h 的数据。
2 结果与分析
2.1 MAN1A1 或MAN1A2 的单敲除细胞的构建
当哺乳动物细胞表达糖蛋白时由于发生于内质网和高尔基体的糖基化过程会产生在蛋白质上糖链的异质性。 糖链的异质性是一个医药用糖蛋白生产中的一个重大的问题。 为了使糖链均一化,我们选择使用人胚胎肾细胞HEK293 用于如病毒的生产和表达重组蛋白质。 由于该细胞株为频繁使用的人源细胞株,它具有如染色体数目和核苷酸多态性等完备的基因组信息,同时积累了大量应用基因表达时的数据。 同时补充说明,在如中国仓鼠卵巢细胞(CHO)中已经建立了悬浮培养和无血清培养的方法,这些在构建表达糖链均一化的重组蛋白质细胞株中很有价值[27-28]。
为了构建生产Man9 和Man8 为主要糖链结构的高甘露糖型糖链的细胞株,我们着眼于定位在高尔基体的甘露糖苷酶Ⅰ。 大多数N-糖基化的蛋白质从内质网转运至高尔基体时, 其主要N-糖链结构为Man9GlcNAc2 或Man8GlcNAc2 的结构。 在高尔基体中,1,2-连接的甘露糖会被高尔基体甘露糖苷酶Ⅰ酶切至Man5GlcNAc2 结构[29-30]。 在哺乳动物细胞中,存在3 种编码高尔基体甘露糖苷酶Ⅰ的基因,MAN1A1、MAN1A2 和MAN1C1。 我们尝试通过CRISPR/Cas9 系统在HEK293 细胞中敲除MAN1A1和MAN1A2 基因。 对于每一个基因的敲除,我们在同一个外显子上设计了两个目标序列。 当敲除质粒转染至细胞,单克隆细胞被分离,单克隆细胞在外显子区域的序列将会被分析,以确定基因是否成功敲除。 成功敲除基因的细胞在设计外显子区域,两个目标序列之间的DNA 片段将会被移除, 我们筛选所有染色体在该区域的DNA 片段均被移除的细胞株。 细胞株A1-KO24 和A2-KO37 被认为是MAN1A1-KO 和MAN1A2-KO 的单敲除细胞株。 相较于MAN1A1 基因序列,图3 展示了对于单敲除细胞株MAN1A1-24 的测序结果,A1-KO24 细胞株在两个目标序列之间存在一个62 bp 的移除。 Cas9 蛋白质正确地执行了对于前间区序列临近基序(PAM)上游3 bp 处目标序列的移除,同时切口被非同源末端连接。 同时62 bp 的移除会造成MAN1A1基因内的移码突变。 同理, 图4 为单敲除细胞株MAN1A2-KO37 的测序结果,A2-37 细胞株中也存在两个目标序列间的32 bp 的移除, 同时这个移除也会造成移码突变。 这些结果显示MAN1A1 和MAN1A2 成功地被敲除了。
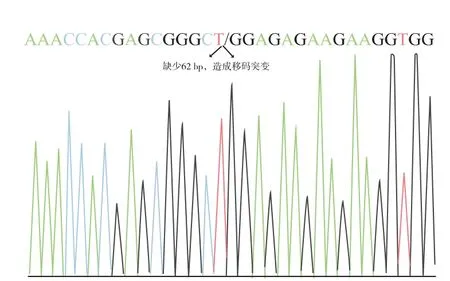
图3 MAN1A1-KO24 单敲除细胞株的性状Fig. 3 Genotype of MAN1A1-KO24 cells

图4 MAN1A2-KO37 单敲除细胞株的性状Fig. 4 Genotype of MAN1A2-KO37 cells
2.2 构建MAN1A1 和MAN1A2 的双敲除细胞株
作者构建了MAN1A1 和MAN1A2 双敲除细胞株(D-KO)。 以A1-KO24 细胞株为亲代,在该细胞株上敲除MAN1A2 基因。 在转染敲除质粒后,细胞被分离。 在敲除目标序列附近的序列被测序分析,用来验证细胞株MAN1A2 被敲除。 对比A2-KO37细胞株, 双敲细胞株double-KO 35 (D-KO35)在MAN1A2 目标序列片段被测序后显示具有3 条不同的结果。 结果如下: 野生型细胞中设计的MAN1A2 靶点之间的序列为aattCCACATGTAGATG CCGGTAAAGGggctaaaaaCCC CGGAGTCTTCCT; 最小的片段是因为两目标序列之间的DNA 片段被移除后裸露端重新连接而形成的aattCCACATAGTCT TCT,存在32 bp 的移除;中间的片段为在目标靶点1 切除位点处插入了75 bp 长度来自于敲除质粒的序列,75 bp 的序列为来自敲除质粒上的氨苄抗性基因片段的TGGTATGGCTTCATTCAGCTCCGGTTC CCAACGAT CAAGGCGAGTTACATGATCCCCCATG TTGTGCAAA AAAGC,同时在目标靶点2 的切除位点处有一个1 bp 的插入碱基;第3 种片段是由于目标靶点1、2 的切除位点处存在2 bp(GA)和207 bp的插入片段,同样来自于敲除质粒的207 bp 的序列为AATAAAAATCATTAGGGGATTCATCAGGGCTTTG CGTTGTTTAATATCCTGATCCTGTCCGGGCGTCGG GCGACCTTTGGTCGCCCGGCCTCAGTGAGCGAGC GAGCGCGCAGAGAGGGAGTGGCCAACTCCATCA CTAGGGGTTCCTGCGGCCGCTCCCCAGCATGCCTG CTATTCTCTTCCCAATCCTCCCCCTTGCTGTCCTGC。因为3 种情况均造成移码突变, 所以可以认为DKO35 为MAN1A1 和MAN1A2 的双敲除细胞株。
2.3 野生型细胞WT、 双敲除细胞D-KO35 的生长情况的测定
在获得了双敲除细胞后,由于破坏高尔基体α-1,2-甘露糖苷酶Ⅰ需要比较其是否对细胞生长状态产生影响,同时也要确认获得的细胞株能否适应生产的需要,需要对细胞的生长情况予以测定。 我们采用CCK-kit[26]对细胞的生长状态进行测定,见图5。从图5 可以看出,细胞增殖速率D-KO 细胞要低于WT 细胞。 说明通过对细胞高尔基体内α1,2-甘露糖苷酶Ⅰ的破坏,会影响细胞增殖。 同时由于敲除α-1,2-甘露糖苷酶Ⅰ的基因对于细胞生长速率产生了影响, 我们也考虑之后通过其他手段,如过表达管家基因等方式实现细胞生长速率的上升。

图5 野生型细胞和D-KO 细胞的生长曲线Fig. 5 Cell proliferation of WT and D-KO cells
2.4 通过流式分析对双敲除细胞表面N-糖链结构变化进行比较
为了验证D-KO 细胞株蛋白质上糖链结构的变化,我们使用多种凝集素来验证该变化。 凝集素是来源于植物、无脊椎动物等的一类糖蛋白,具有凝集血红蛋白的作用。 凝集素具有识别糖蛋白上特定结构的能力,因此在糖生物学研究中往往使用凝集素来验证蛋白质上糖链的结构。 我们使用表1 中的凝集素来与细胞表面蛋白质反应,由于这些凝集素带有异硫氰酸荧光素(FITC)标记,当凝集素与相应识别的目标糖链结构结合时, 带有FITC 的荧光标记可以帮助我们检测到荧光变化,从而说明蛋白质上具有可与该凝集素结合的结构。 荧光的变化通过流失细胞分析仪进行检测。 通过所得到的结果可以计算样品与凝集素的结合情况,即对应于流失细胞分析仪中的峰的Mean 值。 通过对比野生型WT和D-KO 细胞中Mean 值的相对变化可以得出相对荧光强度, 结果见图6。 图6 纵坐标表示以WT 的Mean 值为单位强度的反映D-KO 细胞株的各凝集素相对荧光强度值。 在图6 中,对比表1 提供的各凝集素对于不同糖链结果的特异性识别数据,可以得出, 在D-KO 细胞相较于野生型,ConA-FITC 的相对荧光强度增加, 证明在D-KO 中高甘露糖型N-糖链有所增加,为野生型的5.02 倍。 同时,PHAL4-FITC、PHA-E4-FITC、ECA-FITC、DSA-FITC 的数据中,D-KO 细胞均具有明显的变化,证明细胞表面的蛋白质上糖链中Galβ1、4-GlcNAc 和β1,4-GlcNAc 的结构有明显的减少, 证明复合型N-糖链结构在D-KO 细胞株中明显减少。

表1 流式分析使用的凝集素以及识别糖链特异性结构信息Table 1 Lectin use for FACS and its target glycan structure
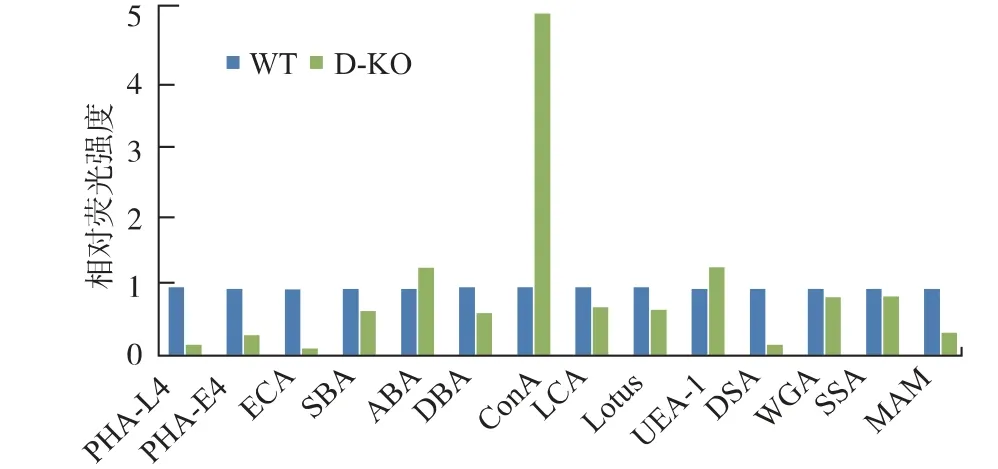
图6 凝集素染色相对荧光强度Fig.6 Lectin staining and the relative fluorescence intensity
2.5 用western bolt 来验证双敲除细胞株内蛋白质上N-糖链结构的变化
在构建了MAN1A1 和MAN1A2 的双敲除细胞株后,我们认为全细胞上的糖蛋白糖链的表型结构已经发生了一定程度的改变。 我们希望通过western blot 的手段来验证糖蛋白上糖链类型的改变, 因此我们选择一种与溶酶体相关的膜蛋白LAMP2 来作为模式蛋白质。 溶酶体相关膜蛋白2(Lysosome-associated membrane protein 2) 也称作CD107b,LAMP2 蛋白质是一种与溶酶体相关的糖蛋白。将细胞用胰蛋白酶消化后,使用PBS 洗涤。在3 000 r/min 下离心3 min 弃去上清液, 将细胞重悬于细胞裂解液中,冰上放置30 min。 将细胞裂解细胞液的样品通过SDS-PAGE 并使用anti-LAMP2 的鼠单克隆抗体识别LAMP2 蛋白质。 在图7 中,野生型和双敲除细胞D-KO 中的LAMP2 蛋白质大小有所不同,表明其蛋白质上糖基化水平存在不同。 通过EndoH 和PNGaseF 处理后的条带变化来看,可以证实这一假设。由于PNGaseF 可以移除几乎所有的糖蛋白上N-糖链, 而EndoH 则可以代谢糖蛋白上高甘露糖型N-糖链。 在野生型中EndoH 处理后与双敲除对比发现,PNGaseF 可以移除的糖链,EndoH不能很好地移除, 对比双敲除细胞,LAMP2 对于EndoH 的敏感程度与WT 明显不同, 进一步表明LAMP2 上糖链表型发生了变化,即高甘露糖型糖链的比例增加,复合型糖链的比例在减少。

图7 LAMP2 的免疫印迹结果Fig. 7 Western blot result of LAMP2
3 结 语
作者构建了一种基于敲除高尔基体α-1,2-甘露糖苷酶Ⅰ控制基因MAN1A1 和MAN1A2 的破坏来实现糖链高甘露糖型N-糖链增加的细胞株。 利用CRISPR/Cas9 系 统, 成 功 敲 除MAN1A1 和MAN1A2 基因,获得双敲除细胞株。 由于细胞内高甘露糖型糖链的增加,使细胞在不同的凝集素染色下,通过流式分析仪分析可以观察到细胞表面蛋白质糖链表型变化。通过对比双敲除细胞株和野生型细胞株的生长状态以及胞内溶酶体膜蛋白LAMP2的糖链变化,可以基本确定双敲除细胞株胞内蛋白质糖链对于EndoH 的敏感性, 即蛋白质糖链为高甘露糖型糖链的证明。 通过使高尔基体甘露糖苷酶基因的两种基因被破坏或敲除,能够获得复合型糖链的含量大大降低、糖蛋白的稳定性和安全性优异的、 以高甘露糖型糖链为主要N-糖链结构的糖蛋白。同时作者也提供了一种用于生产以高甘露糖型糖链为主要N-糖链结构的糖蛋白的动物细胞株、 生产以高甘露糖型糖链为主要N-糖链结构的糖蛋白的方法、通过该方法制备的糖蛋白以及该糖蛋白的用途。
