酯交换法合成碳酸甲乙酯研究进展
2019-12-19王吉宇张志刚许光文
石 磊, 于 悦, 王吉宇, 张志刚, 许光文
(1. 沈阳化工大学 资源化工与材料教育部重点实验室, 辽宁 沈阳 110142;2. 沈阳化工大学 能源与化工产业技术研究院, 辽宁 沈阳 110142;3. 沈阳化工大学 应用化学学院, 辽宁 沈阳 110142)
碳酸甲乙酯(EMC)是一种具有不对称结构的线型碳酸酯类化合物,它同时兼有碳酸二甲酯(DMC)与碳酸二乙酯(DEC)的特性,可作为羰基化或烷基化试剂用于有机合成[1]反应。EMC是一种有前途的汽油添加剂[2],可提升汽油辛烷值[3],降低汽车尾气中固体颗粒物以及氮氧化合物的排放。EMC更为重要和广泛的应用是作为一种优良的锂离子电池电解液溶剂[4],在结构上具有空间位阻小和不对称性,能够辅助增加锂离子的溶解度,提高电池的电容量密度和电量[5];EMC作为溶剂有利于拓宽电解液工作温度范围[6],同时具有优良的导热性、低电阻性和电化学环境结构稳定性[7],有效提升锂电池的安全性[5]并延长电池使用寿命[7,8]。
EMC的合成方法主要有光气法、氧化羰基化法以及酯交换法等[9]。光气法生产效率高,但原料光气剧毒[10,11],产生强酸性HCl严重腐蚀设备,该方法已经逐步被淘汰。氧化羰化法[12]以MeOH、EtOH、CO和O2为主要原料,但是EMC收率太低,且反应有O2参与,存在安全隐患。改进的氧化羰化法中,CO2可以代替CO和O2一步合成碳酸酯[13,14],但碳酸酯收率一般低于20%。酯交换法制备EMC包括DMC与EtOH[15-24]的酯醇交换和DMC与DEC[25-32]的酯酯交换。它们的反应过程如方程式(1)和(2)所示。酯交换法反应条件温和、催化效率高、工艺相对简单、且几乎没有副产物生成,比起其他合成路线更加绿色环保,所以工业上多使用酯醇交换法制备EMC。但该反应体系存在三种共沸产物,分别为MeOH-DMC(63.7 ℃,86.6 mol% MeOH)、EtOH-DMC(74.9 ℃,69 mol% EtOH)和EtOH-EMC(78.9 ℃,95 mol% EtOH)[15],这些共沸物给后续分离带来困难。如果碳酸酯产品中混有少量醇,会造成锂电池效率降低,甚至形成安全隐患。工业上采用催化精馏的方式实现产物分离,主要以强碱性甲醇钠(CH3ONa)为催化剂,催化活性较高,但CH3ONa遇水敏感,容易失活,参与反应生成的钠盐在碳酸酯中溶解度较低,容易析出,导致催化剂不能循环使用,并且难以分离,少量残留就导致生产的EMC纯度降低,产生强碱性固废污染环境。强碱性催化剂也可以催化DMC与DEC的酯酯交换合成EMC,该反应体系不存在共沸分离问题,但是催化剂在碳酸酯中的溶解度较低,工业上暂时无法提供万吨级的DEC。
CH3OCOOCH3+C2H5OH→ CH3OCOOC2H5+CH3OH
(1)
CH3OCOOCH3+C2H5OCOOC2H5→
2CH3OCOOC2H5
(2)
DMC与EtOH的酯交换反应体系中,还包括EMC与EtOH酯交换生成DEC和MeOH、EMC歧化反应生成DMC和DEC、DMC与EtOH酯交换生成DEC和MeOH等三个反应,如方程式(3)、(4)和(5)所示。反应(1)、(3)与反应(5)为相关反应,反应(2)是反应(4)的逆反应。
CH3OCOOC2H5+C2H5OH→
C2H5OCOOC2H5+CH3OH
(3)
2CH3OCOOC2H5→
CH3OCOOCH3+C2H5OCOOC2H5
(4)
CH3OCOOCH3+2C2H5OH→
C2H5OCOOC2H5+2CH3OH
(5)
最新碳酸酯相关综述为2016年张旭[33]发表,主要讨论了DMC酯交换反应合成EMC的路线,对比DMC分别与EtOH和DEC进行反应的特点以及部分催化剂研究进展。本研究在此基础上总结了近五年来该领域的研究新进展,系统整理和综述了酯交换法制备EMC的反应热力学、动力学、催化剂进展、反应机理及反应工艺。并分类阐述了均相催化剂(可溶碱、离子液体和可溶盐类)以及非均相催化剂(离子交换树脂、金属氧化物、碱性分子筛和MOFs材料类)的最新研究,最后指出合成锂电池级EMC所面临的问题以及未来的研究方向。
1 反应热力学


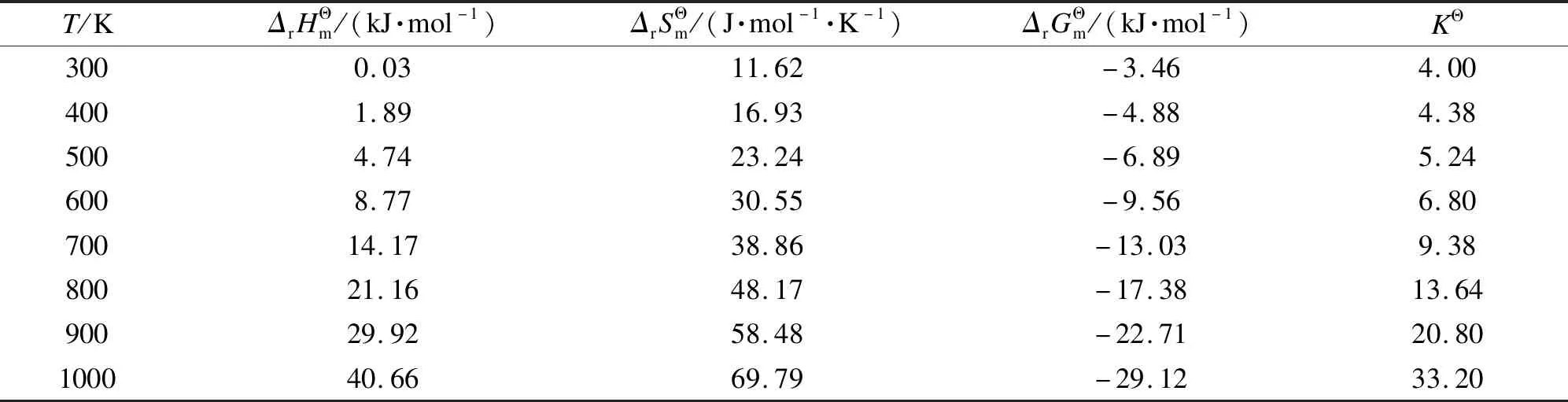
表 1 DMC与DEC酯交换反应的热力学参数[34]
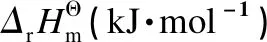
表 2 DMC与EtOH的酯交换反应体系的和

表 3 DMC与EtOH酯交换反应体系的和KΘ[37]
值得注意的是,文献[37]中提到的EMC歧化反应是DMC与DEC酯交换反应[34]的逆反应,两者对各自的讨论对象都得出是吸热反应以及熵增的结论,因此是互相矛盾的,但是对反应自发性的判断是一致的。说明在分析过程中,选择数据处理模型以及引用热力学参数都会对理论分析结果产生影响。针对DMC和DEC酯交换制备EMC的反应,作者完成了20 kg级别固体催化剂中试,催化剂为圆柱形,长度8-12 mm,直径1.5 mm,混合原料中DMC和DEC物质的量比1∶1,混料质量空速5 h-1,原料经预热器加热到105 ℃以后通过单管催化剂床层,反应器已做外保温处理,并在距反应器入口1/3及出口1/3处设有测温热电偶。随着反应进行,反应温度逐渐降低,反应器出口温度在80-85 ℃,产物组成(质量分数)约为19.8% DMC、51.7% EMC以及28.5% DEC,没有其他副产物生成。根据各测温点物料的温度变化情况,得出DMC和DEC酯交换制备EMC是一个吸热反应的事实。

2 反应动力学
通过碳酸酯交换体系中反应动力学研究可以计算出反应速率随温度、反应物浓度随时间变化的规律,借此对EMC的生产工艺进行校验,从而有助于提高生产效率和产品质量。Luo等[15]以K2CO3和相转移试剂为催化剂,基于各物质摩尔浓度计算出反应(1)和(3)在45-100 ℃反应速率常数k(1)和k(3)分别随温度T变化的关系式,即
k(1)=2.148×104exp
(6)
k(3)=3.076×103exp
(7)
由上式可知,反应(3)的活化能比(1)的活化能略小,但是其Arrhenius方程的指前因子比后者低近一个数量级,因此反应(1)反应速率快于(3)。
Zielinska-Nadolska等[16]考察了分子筛、离子交换树脂、水滑石、金属氧化物、改性K2CO3等物质催化DMC与EtOH酯交换反应的动力学特征。反应温度313-348 K,计算出改性K2CO3、凝胶型强酸离子交换树脂(Lewatit K1221)和超强酸离子交换树脂(Nafion SAC-13)催化上述反应的动力学数据如表4所示。由于活性中心的酸碱性差异,反应(1)和(3)体现出不同的动力学特征。改性K2CO3作催化剂时,反应(1)的活化能大于反应(3)的活化能,但是其Arrhenius方程的指前因子数值大约是后者的四倍,因此反应(1)快于(3),此特点与其他文献[15]给出的结果类似;树脂酸催化时,反应(1)的活化能小于反应(3)的活化能,其Arrhenius方程的指前因子明显小于后者。因此,无论是碱性还是酸性催化剂,反应(3)总是这一“连串反应”的控速步骤[16]。

表 4 反应(1)和(3)的Arrhenius方程的指前因子与活化能[16]
Keller等[20]以C2H5ONa为催化剂,基于反应物活度讨论了DMC与EtOH酯交换动力学特性。反应温度323-363 K,反应(1)和(3)的Arrhenius方程为:
(8)
(9)
此结果表明反应(3)的活化能明显大于反应(1)的活化能,两者的Arrhenius方程的指前因子分别为k0,(1)=1.34×1011mol/(m3·s)和k0,(3)=1.80×1013mol/(m3·s),反应速率(1)仍高于(3)。
王红星等[38]考察了303-333 K 时CH3ONa催化DMC与EtOH酯交换体系,建立了基于摩尔浓度的连串反应(1)和(3)的反应动力学方程:

(10)
(11)
式中,cDMC、cEtOH、cEMC、cMeOH、cDEC分别是DMC、EtOH、EMC、MeOH和DEC的摩尔浓度(mol/L)。反应(3)的活化能明显大于反应(1)的活化能,其Arrhenius方程的指前因子大约是后者的4.2倍,反应(1)和(3)的活化能与指前因子的大小关系与文献[20]一致。
张运茂等[39]考察了碱性咪唑离子液体催化DMC与DEC的酯交换特性,该反应是EMC歧化反应的逆反应,测得的均相可逆反应动力学参数见表5。正向反应(2)的活化能大于歧化反应(4)的活化能,但是(2)的指前因子是(4)的74.5倍,因此,反应(2)平衡常数更易受温度影响,提高温度促进反应向正向进行,当反应温度由353.15 K升高到368.15 K时,反应平衡常数由3.04提高至3.45。

表 5 反应(2)和(4)的Arrhenius方程的指前因子与活化能[39]
对于DMC与EtOH酯交换反应体系的动力学研究还存在少许疑问:当催化剂为强碱或酸时,第一步酯交换反应(1)的活化能和指前因子都比第二步酯交换反应(3)的小。但是,以K2CO3为催化剂时,反应(1)的活化能和指前因子皆大于(3),这说明不同碱强度的催化剂催化反应机理可能有所差别。
3 催化剂进展
酯交换法合成EMC的催化剂分均相和非均相两大类,其中,均相催化剂主要包括:可溶有机强碱,如甲醇钠(CH3ONa)、乙醇钠(C2H5ONa)或叔丁醇钠(C4H9ONa)等;可溶无机强碱包括KOH、NaOH等;中等强度碱或弱碱,如K2CO3、Na2CO3或KF等;离子液体,目前研究主要集中在具有不同阴、阳离子结构的烷基咪唑盐,如[Mmim]Cl、[Emim]Br或[Bmim]I等;可溶盐类催化剂,如Mg(NO3)2、La(NO3)3、 Ti(OBu)4或Bu2SnO等。非均相催化剂主要包括:离子交换树脂;金属氧化物,如MgO、CaO、MgO-Al2O3、Mg-Al-O-t-Bu或AlPO等;碱性分子筛,如Al-Zn-MCM-41;MOFs材料,如ZIF-8或ZIF-67等。
3.1 均相催化剂
3.1.1 可溶碱类催化剂
可溶碱类催化剂对酯交换法制备EMC催化效率较高,表6按照催化剂碱强度比较了有机强碱催化剂C4H9ONa、C2H5ONa以及CH3ONa;无机强碱催化剂KOH和NaOH;无机中等强度碱和弱碱催化剂K2CO3、Na2CO3、NaHCO3、KF和NaF对酯交换反应的影响。为保证反应数据的系统性和反应结果的可对照性,本论文进行了部分补充,在参考文献处用“*”来表示。补充数据统一反应条件为:nDMC∶nEtOH=1∶1,催化剂含量0.15%-1.5%(占反应物总摩尔百分数),反应温度30-78 ℃,常压反应0.5 h。
从表6可以看出,有机强碱C4H9ONa、C2H5ONa和CH3ONa作催化剂时,DMC转化率和EMC收率依次下降,表明随着催化剂亲核性减弱,酯交换效率降低。当CH3ONa含量为1.5%,在接近室温(30 ℃)反应0.5 h,取得了良好的催化效果。继续升温至50或者78 ℃,基本保持不变,表明该结果是等物质的量比的DMC和EtOH混合原料在一定温度范围内(0-80 ℃)的平衡转化率。中国科学院成都有机所[40]报道了CH3ONa和CH3OK催化剂在78 ℃反应4 h时的结果,按EtOH计算EMC收率达到54.7%。Keller等[20,21]采用C2H5ONa催化剂用于DMC与EtOH反应精馏过程。有机强碱催化剂如CH3ONa在万吨级/年EMC工业生产过程中会造成精馏塔结垢,产生白膜,同时催化剂在第一精馏塔塔釜析出。姚洁等[40,41]认为,该现象是CH3ONa在反应混合产物中溶解度不好所致,在塔釜中尤为明显。因此,向CH3ONa中添加了醇胺类[41]化合物作溶剂和助催化剂,复合物在反应液中溶解性良好,有利于酯交换反应在均相状下进行,达到了良好的催化效果。
作者发现有机强碱催化剂CH3ONa和无机强碱催化剂NaOH在催化DMC与EtOH的酯交换过程中因参与反应而逐步失活,其反应路径和机理如图1所示:CH3ONa首先与原料中极少量的水(H2O)发生反应,生成NaOH和CH3OH;第二步:OH-会亲核进攻DMC中的羰基碳,使C=O键断裂形成中间体a;第三步:由于a不稳定,与C相连的四个C-O至少需要断裂一条,这是典型的亲核加成-消去反应。1位C-O键在碱催化体系下无法断裂,除非C+和O-能够分别以自由基的形式稳定存在,这显然无法实现。如果3位C-O键断裂则又生成DMC和OH-,2位或者4位键断裂则生成CH3O-和中间体b甲氧基甲酸;第四步:在碱性环境下b无法稳定存在,与NaOH发生中和反应,生成甲氧基甲酸钠和H2O;第五步:OH-还可以亲核进攻甲氧基甲酸钠中的羰基碳,使C=O键断裂形成中间体c;第六步:中间体c中2位C-O键断裂形成CH3O-和NaHCO3;第七步:NaHCO3可以继续与NaOH反应生成Na2CO3和H2O。在第四和七步中生成的H2O会持续参与循环,同CH3ONa反应,直至全部生成催化活性较低的Na2CO3或NaHCO3,导致催化剂失活。失活产物甲氧基甲酸钠、NaHCO3或Na2CO3定性和定量分析会在今后工作中给出详细的表征和证明。万吨级/年EMC合成装置的精馏塔内壁产生白膜是因为CH3ONa失活生成了Na2CO3,Na2CO3无论在碳酸酯还是在MeOH或EtOH溶剂中溶解度均极低,析出并附着在精馏塔内壁。大量累积的Na2CO3导致精馏过程中EMC纯度达不到要求,因此,精馏塔需要每隔一段时间水洗溶解Na2CO3。

表 6 可溶碱类催化剂对酯交换反应的影响
*: this research

图 1 CH3ONa/NaOH催化酯交换反应失活机理示意图
KOH和NaOH是最常用的两种无机强碱催化剂,NaOH碱强度略低,在50 ℃反应时,其催化效率约为KOH的2/3。由于两种碱的碱强度较高,高温下酯交换效率基本一致,均在短时间内达到反应平衡[40]。随着碱强度进一步降低,Na2CO3、NaHCO3和NaF三种碱在50 ℃下对酯交换反应几乎没有活性。李琳等[1]以K2CO3为催化剂,nDMC∶nEtOH=1∶1,100 ℃反应7 h,以EtOH计算的EMC收率为55.7%。加入聚乙二醇(PEG)对K2CO3进行改性[16],nDMC∶nEtOH=1∶2,75 ℃时实现EMC收率51.8%。文献中并没有明确给出PEG增效原因,K2CO3或Na2CO3在碳酸酯、MeOH和EtOH中溶解度极低是客观事实,猜测PEG改性能够提升催化剂与原料的接触效率,增加K2CO3的分散度[42],进而提升催化效率。
可溶碱类催化剂(包括有机碱和无机碱)对DMC和EtOH的酯醇以及DMC和DEC的酯酯交换反应遵循亲核加成-消去反应机理,一般碱强度越高,其亲核性越强,催化效率越高。作者试图以pKb值[43](碱度系数)为标准建立碱强度与催化效率之间的关系。以C4H9ONa、CH3ONa、KOH、NaOH、Na2CO3及NaF为例,其pKb值分别为-5.00、-1.54、-1.10、-0.56、3.60、10.86,pKb值越低其碱强度越强,催化效率越高。使用同种催化剂时,在一定范围内,催化剂加入量越多,催化活性越高;升高反应温度及延长反应时间,均有利于促进反应向正方向进行,提升产物EMC收率,直至反应达到热力学平衡值。
3.1.2 离子液体类催化剂
离子液体具有蒸气压低、溶解性强、热稳定性好、可重复利用等特点[44-46],因此,受到广泛关注,目前,EMC合成的研究主要集中在烷基咪唑盐类离子液体[47-53]。表7考察了具有不同阴、阳离子结构的咪唑盐类离子液体:1,3-二甲基咪唑氯盐([Mmim]Cl)、1-乙基-3-甲基咪唑氯盐([Emim]Cl)、1-丁基-3-甲基咪唑氯盐([Bmim]Cl);1-乙基-3-甲基咪唑溴盐([Emim]Br)、1-丁基-3-甲基咪唑溴盐([Bmim]Br)、1-丁基-2-乙基-3-甲基咪唑溴盐[Bemim]Br、1-丁基-2-丙基-3-甲基咪唑溴盐([Bpmim]Br);1,3-二甲基咪唑碘盐([Mmim]I)、1-乙基-3-甲基咪唑碘盐([Emim]I)、1-丁基-3-甲基咪唑碘盐([Bmim]I);1-丁基-3-甲基咪唑丁酸盐([Bmim]CH3(CH2)2COO)和(1-(4-羟基)丁基-3-甲基咪唑苯甲酸盐([OHBmim]PhCOO)对碳酸酯酯交换反应的影响。

表 7 离子液体类催化剂对酯交换反应的影响
结合本实验室[47]工作分析发现,当阳离子相同时([Bmim]+),阴离子活性变化顺序:I-> Br-> Cl-;阳离子为[Emim]+和[Mmim]+时,阴离子也符合如上规律。原因是卤素阴离子中I-半径最大,较容易摆脱咪唑阳离子的束缚,有利于活化EtOH,增加体系中EtO-的含量,从而提高DMC转化率和EMC选择性。亓虎等[48]合成了[Bmim]Br离子液体,反应温度由90 ℃升至140 ℃,DMC转化率提高9.4%,EMC收率提高13%[49]。说明在一定范围内升高温度有利于提高反应物分子之间的有效碰撞,促进反应正向进行,从而提高反应物转化率及产物收率。李永昕课题组[50,51]合成了[Bemim]Br和[Bpmim]Br两种离子液体分别催化DMC与DEC以及DEC与MeOH的酯交换反应,在相对较高温度长时间反应才得到60%EMC收率。[Bmim][CH3(CH2)2COO][52]催化双酯交换反应,增加DMC含量,DEC转化率升高,催化剂重复使用五次后仍保持较高的转化率,合成的[OHBmim]PhCOO[53]离子液体重复使用六次活性没有变化。
咪唑类离子液体在催化EMC合成过程中受烷基取代基、卤素和反应温度等因素影响较为明显。当阳离子相同时,阴离子活性变化顺序为:I-> Br-> Cl-;阴离子相同时,阳离子活性变化顺序为:[Mmim]+> [Emim]+> [Bmim]+;酯交换反应制备EMC是一个可逆反应,在一定范围内,增加一种反应物浓度、升高反应温度,均有利于反应向产物方向进行,提升EMC收率。
3.1.3 可溶盐类催化剂
Mei等[18,54]研究了不同种硝酸盐催化剂用于DMC与EtOH的酯交换反应效果,催化活性顺序为:La(NO3)3> Mg(NO3)2> KNO3> Al(NO3)3,DMC转化率最高可以达到86.5%。文献[54]认为La3+具有较强的Lewis酸性,能够活化反应底物,降低反应活化能。以La(NO3)3·6H2O为催化剂时,同La(NO3)3催化反应结果相差不大,说明以La3+(Lewis酸)为活性中心催化酯交换反应时,受水的影响不大。文章[54]还研究了LaCl3以及La(CF3SO3)3催化效果,DMC转化率分别为48.0%和83.2%,其中,LaCl3催化活性小于La(NO3)3和La(CF3SO3)3,认为这与阴离子同配位中心与La3+间配位作用力大小有关,由于Cl-与La3+之间的配位能力较强,抑制了体系中EtOH、DMC等供电子物质占据配位点,难以活化反应底物,所以催化剂活性相对较差。综上,对于可溶性Lewis酸催化剂,其接受孤对电子能力越强即其酸性越强,其催化酯交换能力越好,反应体系中的水对催化活性影响不大。
卓广澜等[55]以钛酸盐Ti(OBu)4或Ti(OPh)4为催化剂,催化DMC和DEC的酯交换反应,当nDMC∶nDEC=1∶1,催化剂含量为反应原料总量的2.5%,103 ℃反应3 h时DEC转化率分别为43.1%和41.7%。通过GC-MS分析,检测到-OPh或-OBu官能团与-OCH3或-OC2H5发生交换的副产物,导致EMC选择性未能达到100%。通过本实验室经验分析这可能是钛酸盐类均相催化剂的共同缺点,虽然催化效率较高,但钛酸盐与原料进行酯交换或钛酸盐水解会生成副产物。对于碳酸酯的酯交换反应,今后可尝试以Ti(OCH3)4或Ti(OC2H5)4为催化剂,既保证催化剂在碳酸酯中较好的溶解性,良好的催化活性,又没有副产物生成。文献[55]同时考察了有机锡类化合物Bu2SnO和BuSnCl3的催化性能,DEC转化率分别为45.6%和43.3%,产物EMC选择性可以达到100.0%。从实验数据分析,在催化DMC与DEC的酯交换反应中,有机锡类化合物比钛酸盐类催化性能略高。Lewis酸催化酯交换反应在于配位化合物中的金属存在不饱和空轨道,能够接纳羰基O的孤对电子,从而活化C-O-,发生-OCH3和-OC2H5酯交换反应。
3.2 非均相催化剂
3.2.1 离子交换树脂类催化剂
Zielinska-Nadolska等[16]研究了离子交换树脂类催化剂对DMC和EtOH酯交换的影响,反应条件:nDMC∶nEtOH=1∶2,75 ℃反应26.7 h,催化剂占反应原料总质量的6.75%。发现强酸性磺酸树脂中Lewatit K1221催化活性最高,DMC转化率54%,EMC收率53%。文献认为该树脂聚合结构中二乙烯基苯含量最低,导致树脂交联度比较低,在树脂溶胀过程中有更多孔道形成,促进反应原料及产物扩散,提升催化效率。作者更倾向认为制备工艺和厂家不同,导致树脂酸含量、酸强度以及孔道结构不同,影响酯交换反应活性。大孔弱酸性树脂Dowex MAC-3,其酸性官能团为聚丙烯酸和羧酸,该树脂无催化活性;超强酸性树脂Nafion SAC-13为四氟乙烯和全氟-3,6-二氧杂-4-甲基-7-辛烯磺酰氟的共聚物,由于-CF2CF2SO3H的强吸电子能力导致磺酸基团具有超强酸性,其催化活性较高,与Lewatit K1221接近。一般酸性树脂的活性中心为苯磺酸,苯磺酸还可以直接嫁接到其他材料上。薛冰等[56]采用氧化石墨烯和对氯苯磺酸为原料,以共价键的方式将磺酸基嫁接到氧化石墨烯表面,获得一种强酸性固体催化剂。nDMC∶nEtOH=1∶1,90 ℃反应6 h,催化剂占DMC质量的3.0%,EMC收率为86.7%,催化剂重复使用三次,EMC收率仅下降3.6%。
3.2.2 金属氧化物类催化剂
杨延钊等[57]以AlCl3·6H2O和氨水为主要原料,采用沉淀法合成了γ-Al2O3催化剂,在600 ℃焙烧4 h后用于DMC和DEC的酯交换反应。nDMC∶nDEC= 3∶1,100 ℃反应3 h,催化剂加入7.0%,EMC收率约63.0%。这类催化剂的优势是机械强度高,催化活性好。柳娜等[58]采用真空浸渍法制备了Al2O3/SiO2催化剂,当Al2O3负载量12%,催化剂加入量7%,nDMC∶nDEC=1∶1,120 ℃反应8 h时,EMC的收率可以达到63.6%。通过表征手段观测到随着Al2O3负载量的增加,催化剂表面活性位数量随之增加,因此,收率也不断提高;但当Al2O3负载量超过一定量(12%)时,可能会堵塞催化剂部分孔道,传质作用减弱,导致产物收率下降。负载量为12%的Al2O3/SiO2催化剂重复使用三次,EMC收率仅降低1.3%,未发现明显失活,稳定性较好。
Murugan等[19]合成了不同KF负载量的KF/Al2O3催化剂,用于催化DMC和EtOH的酯交换反应。反应条件:nDMC∶nEtOH=1∶4,80 ℃反应4 h,使用无水KF为催化剂时,EMC收率为49.6%,采用10% KF /Al2O3催化剂时,EMC收率为51.4%,但是以20% KX/Al2O3(X为Cl、Br、I中的一种)作催化剂时,均无催化活性。文献认为可能是KF和Al2O3在反应过程中形成表面活性物质Al-[OH…F]-和K+,Al-[OH…F]-物种作为活性中心有利于活化EtOH产生EtO-,EtO-亲核进攻DMC生成EMC,提高了反应效率。此外,分别在300、600以及900 ℃下焙烧20% KF/Al2O3催化剂,发现均形成了氟铝酸钾(K3AlF6),焙烧温度越高,活性组分K+和F-损失越多,催化剂碱强度下降,导致活性降低。
Shen等[25]在气相和液相体系中研究了自制固体碱催化DMC和DEC的酯交换反应,发现催化活性顺序一致:MgO>ZnO>La2O3>CeO2,具有较多强碱性中心的La2O3和较多弱碱性中心的CeO2催化活性均较差,而ZnO和MgO中等碱性位点较多,活性较好,文献认为中等强度的碱性中心更有利于催化DMC和DEC的酯交换反应。同时MgO的BET比表面积大于ZnO,吸附反应物多,反应相对容易进行,催化活性较高。陈英等[59]将市售MgO、CaO于500 ℃焙烧4 h后用于催化酯交换反应,nDMC∶nDEC=1∶1,90 ℃反应时EMC收率均在1%-5%,升温至100 ℃时,EMC收率约为35%和5%,继续升高温度至103 ℃,EMC收率在44%和41%左右,催化效果差别不大。由此可见,反应温度对MgO和CaO两种催化剂活性影响很大。
Zhao等[28]选用不同炭材料作载体,包括杏壳炭(AC)、煤质炭(CC)及介孔CMK-3和NC-2,采用浸渍法制备了负载型MgO催化剂,活性组分MgO负载量为4.0%。反应条件:nDMC∶nDEC=1∶1,103 ℃反应0.5 h,催化剂加入4.8%,MgO/NC-2为催化剂时,EMC收率49.3%,其他三种催化剂的收率均在10%以下,活性顺序为:MgO/NC-2>MgO/CC>MgO/AC>MgO/CMK-3。MgO/NC-2催化剂使用后在氩气气氛中800 ℃焙烧可以再生,重复使用五次其催化活性基本不变。结合表征分析,NC-2材料表面存在着丰富的羧酸、内酯、酸酐等含氧基团,该含氧基团与Mg物种之间相互作用,促使MgO高度分散,进而提高催化活性和稳定性。Lv等[60]以HZSM-5为载体制备了负载型催化剂,采用催化精馏装置进行评价,nDMC∶nEtOH=2.56∶1,90 ℃反应7 h,回流比20∶1,当以HZSM-5、2.3%CaO/HZSM-5和2.3%MgO/HZSM-5为催化剂时(2.3%为MgO相对于HZSM-5的质量百分比),EMC收率分别为46.9%、86.1%和97.5%。结合表征发现,三种催化剂均具有介孔结构,其中,2.3%MgO/HZSM-5催化剂碱量最高,文章认为碱性越强,越有利于活化EtOH形成EtO-,促进酯交换反应的进行。
陈英等[59]采用共沉淀法合成了MgO-Al2O3复合金属氧化物,沉淀剂为NaOH和Na2CO3,发现该催化剂催化活性优于市售的MgO以及CaO。nDMC∶nDEC=1∶1,103 ℃反应4 h,EMC收率可达45.8%。XRD谱图发现MgO-Al2O3复合氧化物中存在MgAl2O4物种,猜测该物种与A12O3晶面协同催化,从而提高催化活性。CO2-TPD分析表明,MgO和CaO催化剂均具有较强的碱中心,而MgO-Al2O3碱性较弱,文章认为较弱的碱性位更有利于催化DMC和DEC的酯交换反应。Wang等[61]以异丙醇铝和硝酸镁为主要原料,通过蒸发诱导自组装法合成介孔镁铝尖晶石MgAl2O4(MAO)催化剂,采用MAO-x表示(x为Mg/Al物质的量比)。nDMC∶nDEC=1∶1,103 ℃反应0.5 h,催化剂加入5%,以MAO-1、MAO-1.5和MAO-2为催化剂时,EMC收率均接近49.0%(达到平衡转化率);以MAO-0.5和MAO-3为催化剂时,EMC收率分别为23.0%和18.0%。结合表征分析,MAO催化剂具有酸碱双功能特性,所有样品表面均存在弱酸性位,酸强度几乎相同,但碱强度存在明显差异。MAO-0.5碱强度最弱,随着Mg含量增加,催化剂碱强度增强,MAO-2具有中等强度碱性位,而MAO-3仅展现出弱碱性位,催化活性也呈现先上升后下降的趋势,因此,得出结论:MAO系列催化剂随着碱强度增强,其催化活性逐步提升。分离MAO-2催化剂,80 ℃干燥5 h后循环利用五次,EMC收率仅降低1.2%,说明催化剂具有良好的重复使用性能。Shi等[29]以硝酸铝为铝源,采用柠檬酸辅助溶胶-凝胶法制备无定型的介孔磷酸铝材料(AlPO),以异丙醇铝和三乙胺为原料通过常规的溶胶-凝胶法合成微孔磷酸铝材料(AlPO-5)。nDMC∶nDEC=1∶1,反应温度93 ℃,5.0%催化剂,活性顺序为:AlPO>MgO>Al2O3>AlPO-5>P2O5,AlPO仅反应0.5 h即实现47.7% DEC转化率(平衡转化率)。AlPO重复使用三次后,其催化活性下降2%左右。表征发现AlPO材料表面存在较丰富的弱酸和弱碱中心,有利于活化DMC和DEC;而且介孔材料的孔径较大,促进了反应原料及产物间的传质,提升了反应速率。Chen等[62]合成了一种酸碱双功能介孔材料MgO-Al2O3-SBA-15(SBA-15为介孔二氧化硅),简写为MA-SBA-15(n),n表示MgO-Al2O3的质量百分比。nDMC∶nDEC=1∶1,104 ℃反应4 h,催化剂加入4%,MA-SBA-15(10)为催化剂时,EMC收率低于30%;随着镁铝物种含量增加,EMC收率不断提高,其中,MA-SBA-15(30)活性最佳,EMC收率达到46.1%,比SBA-15高20余倍;继续增加镁铝物种含量至40%,此时复合材料的介孔结构被破坏,催化效率有所下降(约44% EMC收率)。通过表征发现未经改性的SBA-15仅有较少量的酸碱活性位,而所有的MA-SBA-15都存在较多量的酸碱活性位。随着镁铝物种含量增加,催化剂酸碱活性位数量也逐渐增多,催化活性加强,但达到一定值(40%)时,会破坏催化剂介孔结构,影响物质间的传质作用,导致催化活性减弱。适量的酸碱活性位及优良的介孔结构共同作用,有利于提升催化效率。
Wang等[32]通过共沉淀法制备磁性Mg-Fe二元复合氧化物催化剂,采用xMgFe-t表示(x为Mg/Fe物质的量比,t为焙烧温度)。nDMC∶nDEC=1∶1,100 ℃反应1.5 h,催化剂加入量1%,MgFe-400催化剂能够实现51%EMC产物收率。XRD和Mossbauer光谱表明,催化剂中Mg与Fe氧化物之间存在协同效应,有助于提高催化剂性能。催化剂在400 ℃焙烧时活性最高,文章认为随着焙烧温度升高,催化剂中MgO含量降低,总碱量减少,导致EMC的收率显著下降。MgFe-400催化剂循环使用10次,催化性能保持不变。
Miao等[63]采用“一锅法”合成双金属Co/Zn-ZIF前驱体(ZIF为沸石咪唑酯骨架),然后在N2气氛下热解得到ZnCo/NC(NC是ZIF的分解产物)催化剂。nDMC∶nDEC=1∶1,100 ℃反应7 h,催化剂加入量1.0%。采用ZnO/NC-600(600为催化剂煅烧温度)作催化剂时,EMC收率为29.5%;以ZnCo/NC-600为催化剂时,EMC收率为51.5%,催化效率接近前者的两倍,结合CO2-TPD表征结果,文献认为Zn、Co两种元素间的相互作用促进了强碱性活性位的产生,更有效地促进了酯交换反应。该催化剂重复使用五次,EMC收率下降11.3%。同时发现600 ℃煅烧的催化剂比550、650、700 ℃煅烧的催化剂拥有更高的活性。根据XRD谱图分析,当煅烧温度为600 ℃时,前驱体中的Zn元素变为ZnO晶体,高于或低于600 ℃煅烧均未形成该结构,说明ZnO对于Zn-Co材料的催化活性是必不可少的,煅烧温度影响催化剂的微观结构和化学状态,进而影响催化性能。作者更倾向于认为双酯交换的催化活性中心为ZnO和表面官能团丰富的炭材料,当焙烧温度较低时,Co/Zn-ZIF没有完全分解,并未形成ZnO活性中心,起到催化作用的是炭材料表面官能团;当焙烧温度适中时,形成了ZnO活性中心,但ZnO在高温条件下升华,因此,继续升高温度时,ZnO升华导致催化剂中ZnO含量较低,催化效果较差。
Mei等[17]以NaOH为沉淀剂,硝酸盐为前驱体,采用共沉淀法制备了Mg-Al-NO3HT催化剂;然后将Mg-Al-NO3HT在叔丁醇钾的四氢呋喃溶液中搅拌24 h制备Mg-Al-O-t-BuHT催化剂;通过尿素水解法制备了Mg-Al-CO3HT催化剂;将Mg-Al-CO3HT在500 ℃焙烧5 h后得到Mg-Al HT催化剂。nDMC∶nEtOH=1∶5,80 ℃反应7 h,催化剂加入1%,以Mg-Al-O-t-Bu HT为催化剂时,DMC转化率可达86.4%,EMC选择性25.9%,循环使用五次催化效果基本不变,其他催化剂活性顺序为:Mg-Al-NO3HT>Mg-Al-CO3HT>Mg-Al HT。Mg-Al-O-t-Bu HT催化DMC与EtOH反应生成的主产物是DEC,其他催化剂主产物为EMC,说明Mg-Al-O-t-Bu HT催化酯交换效率更高。Srivastava等[64]以亚铁氰化钾、氯化锌和叔丁醇以及三嵌段共聚物EO20PO70EO20为原料合成Fe-Zn双金属氰化物配合物,nDMC∶nEtOH=1∶10,反应釜中170 ℃反应8 h,催化剂加入5%,DMC转化率接近98%,EMC选择性接近38%。相比较而言,Fe-Zn双金属氰化物配合物催化酯交换效率不高。综上,双金属复合氧化物催化酯交换反应活性明显优于单一金属氧化物,归功于酸碱协同作用,并随着碱性的增强,其催化剂效率逐渐提升。
3.2.3 碱性分子筛类催化剂
Palani等[26]以偏硅酸钠为硅源、硫酸铝和硫酸锌为主要原料,通过水热法合成了一系列MCM-41分子筛催化剂(Si/Al=50,Si/(Al+Zn)=100)。固定床评价,nDMC∶nDEC=1∶1,原料流量1.5 mL/h,混合原料质量空速约为3.0 h-1,反应温度175 ℃,发现Al-Zn-MCM-41(50)活性最佳,EMC收率85%,反应活性与吡啶红外测定的催化剂B酸含量一致:Al-Zn-MCM-41(50)> Al-Zn-MCM-41(100)> Al-MCM-41(50)> Al-MCM-41(100)。随着反应温度从125升至175 ℃,EMC收率逐渐增加,但温度达到200 ℃时,EMC收率下降,归因于碳酸酯在高温下分解。
3.2.4 MOFs材料类催化剂
Zhou等[27]将六水合硝酸锌和对苯二甲酸溶解于N,N-二甲基甲酰胺中,再加入有机碱三乙胺形成沉淀,洗涤干燥后制得金属有机骨架MOF-5催化剂(Zn4(O)(BDC)3,BCD为苯-1,4-二羧酸酯)。nDMC∶nDEC=1∶1,100 ℃反应3 h,催化剂加入2%:合成原料六水合硝酸锌基本无催化效果,MOF-5催化剂展现出50.1%EMC收率。通过表征发现,MOF-5催化剂在90或300 ℃下干燥相同时间,催化剂结构及表面官能团保持不变,说明MOF-5热稳定性较好。重复使用三次后,EMC收率仅降低1.7%。Zhou等[30]以2-甲基咪唑、氨水和Zn(OH)2为主要原料合成沸石咪唑骨架ZIF-8([Zn(MeIm)2],MeIm为2-甲基咪唑)。nDMC∶nDEC=1∶1,100 ℃反应3 h,催化剂加入1%:以制备原料Zn(OH)2、2-甲基咪唑或两者的混合物为催化剂时,几乎没有催化活性;MgO催化剂活性一般,EMC收率为30.5%;ZIF-8展现出最优的催化效果,EMC收率为50.7%;同为金属有机骨架结构的MOF-5催化剂活性偏低,EMC收率为42.0%,而且MOF-5在潮湿的空气中易分解,不能稳定存在[65]。BET、CO2-TPD和NH3-TPD表征证明,ZIF-8具有高比表面积及酸碱双功能特性。Yang等[66]采用2-甲基咪唑、甲醇和[Zn(NO3)2]·6H2O为原料制备ZIF-8催化剂,并添加了MS 4A分子筛作为助催化剂。nDMC∶nDEC=1∶1,100 ℃反应12 h,仅以ZIF-8或MS 4A为催化剂时,EMC收率分别为50.32%、34.89%,向ZIF-8中加入助催化剂时,催化活性明显提高,EMC收率达到84.72%。Yang等[31]以2-甲基咪唑和六水合硝酸钴为主要原料合成沸石咪唑骨架ZIF-67(Co(MeIm)2)。nDMC∶nDEC=1∶1,100 ℃反应24 h,催化剂加入2%:采用ZIF-8催化剂时,EMC收率为50.32%;ZIF-67催化活性较高,EMC收率高达83.39%。将ZIF-67重复利用三次后,EMC收率减少2%左右,稳定性较好。结合表征发现ZIF-67酸碱性适中,同时ZIF-67的比表面积大于ZIF-8,文献认为中等强度酸碱性和高比表面积更有利于催化双酯交换反应。Desidery等[24]以均苯三甲酸和ZrOCl2·8H2O为原料制备MOF-808(Zr6O4(OH)4(BTC)2(HCOO)6)酸碱双功能催化剂。nDMC∶nEtOH=1∶3,90 ℃反应24 h,催化剂加入1%,比较了多种MOF材料催化双酯交换的结果,发现Cr-MIL 101和Fe-MIL 100完全没有活性,其他MOF材料活性顺序为:MOF-808>UiO66-NH2>>UiO66-SO3H>ZIF-67>ZIF-8>UiO66,由于MOF-808比表面积、孔体积和孔径较大,能够有效促进产物扩散;大量酸碱性位点的协同催化作用提升了酯交换效率。MOF-808在75 ℃反应24 h也能够达到49.6% EMC收率,循环使用三次后,产物收率仅下降0.8%。
4 反应机理
据作者所知,目前文献报道的DMC与EtOH[19,24]或DMC和DEC[25-27,29,31,32,55]酯交换反应机理是根据有机化学基础理论推导得来,而不是由光谱等表征手段真实观测证明,推断的酯交换反应机理如图2-5所示。图2为亲核试剂烷氧基负离子(CH3O-或C2H5O-)与碳酸酯(DEC或DMC)交换后生成EMC的过程。烷氧基负离子首先进攻DMC或DEC中的羰基,使C=O键断裂形成中间体a或b,a和b结构相近,稳定性均较差,与C相连的四个C-O至少需要断裂一条,这是典型的亲核加成-消去反应过程。顶端C-O-键断裂困难,除非C+和O-能够分别以自由基的形式稳定存在,这显然是无法实现的。其他位置C-O键断裂可生成EMC和CH3O-或C2H5O-。
可见,烷氧基负离子地生成在碳酸酯交换过程中十分重要。几种典型催化剂:醇钠或醇钾有机碱、能够电离出OH-的可溶碱、离子液体和金属氧化物活化甲醇、乙醇、或碳酸酯过程如图3-5所示。醇钠或钾(以CH3ONa或CH3OK为例)在有机溶剂中很容易电离出CH3O-,CH3O-可以进攻并夺取C2H5OH中的H生成CH3OH和C2H5O-;同样C2H5O-可以夺取CH3OH中的H生成C2H5OH和CH3O-。该反应是一个可逆反应,但理论上由于甲基的供电子效应使得C2H5O-电负性比CH3O-强,C2H5O-进攻CH3OH更容易进行。OH-通过氢键与CH3OH分子中的羟基H相互作用,可以活化CH3OH分子,生成CH3O-,另一种产物为H2O,该反应也为可逆反应,逆向进行平衡常数更大,也就是说CH3O-更容易与H2O反应,生成OH-和CH3OH。如果希望提高反应体系中CH3O-的浓度,可以将H2O分离出反应体系,推动反应正向进行。OH-也可以进攻DMC或DEC中的羰基,发生亲核反应,生成带负电的中间体,该中间体解离出CH3O-并生成甲氧基甲酸,甲氧基甲酸在碱催化体系下不能稳定存在,会迅速与OH-作用生成甲氧基甲酸盐与H2O。

图 2 烷氧基负离子活化碳酸酯机理

图 3 无机可溶强碱活化碳酸酯机理

图 4 咪唑类离子液体活化甲醇生成烷氧基负离子过程
以1,3-二甲基咪唑碘盐为代表描述咪唑类离子液体活化醇类化合物(以MeOH为例),生成烷氧基负离子过程如图4所示,卤素负离子I-与CH3OH分子接触并通过诱导作用形成氢键,I-夺走H使羟基中的氢氧键断裂并形成CH3O-。理论上I-也可能直接亲核进攻DMC中的羰基,使C=O键断裂,解离出CH3O-并生成碳酸碘甲酯,但GC-MS联用并未检测到该产物地生成。
根据文献[25,27,29],金属氧化物中的O通过共价键与不同金属相连,同时存在未配对的孤对电子,其活化醇类化合物(以MeOH为例),生成烷氧基负离子过程见图5。O孤对电子显电负性,会吸电子诱导CH3OH中的H,形成类CH3O-结构。O孤对电子也可以亲核进攻DMC中的羰基,使C=O键断裂,形成DMC在金属氧化物表面吸附的物种以及解离出类CH3O-结构。金属氧化物催化碳酸酯交换机理还需要原位表征手段证明。

图 5 金属氧化物活化甲醇生成烷氧基负离子过程
5 反应工艺
以DMC和EtOH为反应原料通过酯醇交换法合成EMC的反应体系中主要包含五种物质:MeOH、EtOH、DMC、EMC和DEC,存在MeOH-DMC(63.7 ℃,86.6% MeOH)、EtOH-DMC(74.9 ℃,69% EtOH)以及EtOH-EMC(78.9 ℃,95% EtOH)[15]三组共沸物。由于酯醇或酯酯间交换均为可逆反应,为了使反应正向进行得更彻底,生成更多的EMC,需打破反应平衡,尽快将产物MeOH移除反应体系。催化精馏技术借助精馏分离与酯交换反应的耦合来强化反应正向进行,从而实现EMC的连续化生产。
闫志军等[67]将混有C2H5ONa催化剂的EtOH原料从反应精馏塔中部泵入,DMC从塔釜进料,VDMC∶VEtOH= 2∶1,反应温度90-110 ℃,塔顶收集MeOH、EtOH和DMC进入轻组分精馏塔,塔釜DMC、EMC和DEC进入重组份精馏塔。轻组分精馏塔操作条件:回流比2-4,塔釜76-78 ℃,塔顶63.7-64.7 ℃,塔顶收集到MeOH和DMC(≥98%)共沸物,再进一步分离DMC,塔釜EtOH(≥95%)重新回到反应精馏塔;重组分精馏分离按三步进行:首先控制塔顶温度低于91 ℃蒸出DMC,然后升至91-106 ℃,收集含有少量DMC的EMC混合物,重新回到重组分精馏塔釜再次循环精馏,最后升至106-110 ℃,从塔顶蒸出EMC产品,塔釜收集DEC,最终EMC总收率为35%。梁志广等[68]设计并优化了生产EMC的工艺流程,如图6 所示。同样以均相C2H5ONa为催化剂,反应物DMC、EtOH分别由C1反应精馏塔中部或下部进料,塔釜混合液经过固液分离后得到粗产品,再由精馏塔C2分离,顶部收集的DMC循环利用,塔釜为EMC和DEC的混合液,再经过下一个精馏塔C3分离收集纯产品EMC和DEC;C1塔顶为MeOH-DMC共沸物,经C4萃取精馏塔(邻二甲苯作萃取剂)脱除MeOH,再经精馏塔C5实现DMC和萃取剂的分离。文中利用化工模拟软件Aspen Plus优化了精馏塔操作参数,总进料流量105 kmol/h,EtOH进料流量40 kmol/h,回流比约1.4,萃取剂85 ℃循环利用,实现联产EMC和DEC,且两种产品纯度均达到99.8%。

图 6 EMC生产工艺流程图[68]
Keller等[21]采用中试规模(高度5.4 m,直径50 mm)的精馏塔对DMC与EtOH的酯交换反应进行研究,反应精馏柱示意图及精馏柱装填如图7和图8所示。以C2H5ONa为催化剂,原料分别于填料高度2.0 m处进料,进料温度60 ℃,通过分子筛吸水减少原料中的含水量,以免C2H5ONa催化剂遇水失活。改变操作参数:EtOH与DMC物质的量比由0.9变为2.6,催化剂由0.1%增至0.2%,回流比从1.2改为2.0,馏出物与进料量质量比由0.27升至0.48,实现由EMC为主产物(79.2%选择性)转变成DEC为主产物(80.2%选择性),证明了反应精馏塔可以作为多功能的反应器完成多种产物选择性调控。Werth等[22]研究了微波辅助对反应精馏的影响,模拟精馏塔总填料高度为5.4 m,其中,2 m作为反应部分,以C2H5ONa为催化剂,nDMC∶nEtOH=1∶1,回流比1.2,馏分与进料比0.35。常规实验在加热套或油浴中进行,微波实验以微波合成仪作为加热源/再沸器,发现微波辅助对反应结果影响较小,没有观察到分离效率明显提高。
Luo等[15]为了使反应原料在精馏塔中停留更长时间,将DMC和EtOH原料由塔釜加入,催化剂K2CO3和助催化剂从塔顶加入,nDMC∶nEtOH=1∶6,回流比6,DMC的进料速率为5.5 mol/h,再沸器温度132.3 ℃,维持精馏塔内部温度80 ℃左右,塔顶温度75.5 ℃,能够实现DMC几乎完全转化,DEC选择性大于99.5%。王红卫等[69]采用间歇精馏装置考察精馏塔操作条件(回流比、塔板数等)对混合物分离效果的影响。原料(0.2% MeOH、0.5% EtOH、9.3% DMC、56% EMC、34% DEC)加入塔釜,然后采取全回流操作,当塔顶、塔釜温度稳定后,再按回流比采出,当理论塔板数50,回流比17.5,投料量与上升蒸气量之比为5 kg/(kg·h-1)时,EMC产品纯度最高能够达到99.5 %。

图 7 Keller使用的中试规模反应精馏柱[21] Figure 7 Scheme of the pilot-scale RD column used by Keller[21]
孙兰义等[70]发明了共沸反应精馏法生产EMC,将DMC、EtOH及CH3ONa催化剂分别加入共沸反应精馏塔,再加入共沸剂正己烷,得到第一混合物(DMC、EMC及DEC)和第二混合物(MeOH和正己烷),第一混合物依次经隔壁塔,分离得到三种碳酸酯产品。第二混合物进入分液罐中,向其中加入水分离出正己烷,并得到第三混合物(MeOH+H2O),再经过提纯塔分离得到纯物质。共沸剂能够抑制MeOH-DMC形成,方便分离,简化工艺流程,降低反应能耗。贾风雷等[71]改进了反应塔的内部结构,采用多级丝网填料体和多级塔盘式填料体,且两者间隔安装排列,实现连续进料产出产品,避免堵塞,解决了丝网填料堵塞降低反应效率的问题,提高装置处理量。对于MeOH-DMC共沸物,目前工业上主要采用萃取精馏法和变压精馏分离法。萃取精馏法是加入一种挥发性小、不与原组分形成新共沸物的萃取剂,以此提高原组分间的相对挥发度,实现共沸物分离[72]。该体系中使用最多的萃取剂有邻二甲苯[73]、乙二醇、糠醛、水、氯苯及N,N-二甲基甲酰胺[74]等。毕利君等[75]和姚林祥等[76]分别采用常压(0.1 MPa)-加压(1.3 MPa)-常压(0.1 MPa)精馏塔和常压(0.1 MPa)-加压(1 MPa)精馏塔分离共沸物,通过模拟软件优化了塔板数、进料位置和回流比,均实现DMC和MeOH质量分数达到99.9%以上。

图 8 流体分离实验室中试精馏柱(a); 填充Sulzer BXTM填料的玻璃段柱(b); PT-100热电偶液体分布器(c); 第一隔离层和加热丝(d); 分子筛(e) [21]Figure 8 Pilot-scale RD column at the laboratory of fluid separations(a); glass segments filled with Sulzer BXTM packing elements(b); liquid distributor with PT-100 thermocouple(c); first isolation layer and heating wire(d); molecular sieves(e)[21]
石磊等[77]发明一种均相耦合非均相催化EMC的生产工艺,图9为流程示意图。原料DMC和EtOH经计量泵P-1泵入K-1预反应釜,加入均相催化剂CH3ONa反应5-10 min即达到热力学平衡值,其中,DMC转化率接近70%,EtOH转化率接近60%,产物中EMC选择性约为80%,DEC选择性为20%。第一混合物进入T-1第一反应精馏塔,回流比2∶1,塔顶温度63.6 ℃采出共沸物(DMC和MeOH),质量比接近3∶7,然后通过高、低压或萃取-精馏分离得到DMC和MeOH。塔釜反应混合物(0.02%MeOH、0.2%EtOH、12%DMC、64%EMC及24%DEC)闪蒸蒸去MeOH及EtOH得到第二混合物,经E-1冷凝器冷凝,F-1过滤器除去不溶的催化剂,然后通入酸性CO2气体,再次过滤后得到的第三混合物(DMC、EMC和DEC)进入T-2第二精馏塔,缓慢升温,当塔顶温度94 ℃时,采出DMC组分,125 ℃时采出EMC组分,其中,纯度合格的作为产品。不合格的EMC、塔顶采出的DMC以及塔釜DEC作为原料,同时额外加入DMC配成物质的量比为1∶1的混合原料。混合原料经过预热器加热到一定温度后进入装有自制固体碱催化剂的B-1固定床反应器,反应温度75-120 ℃,原料质量空速0.2-20 h-1,反应后的产物组成大致为20%DMC,55%EMC以及25%DEC,重新循环至第二精馏塔塔釜分离。

图 9 均相耦合非均相催化高纯EMC合成工艺流程示意图[79]
6 结 论
本研究介绍了酯交换法合成EMC的反应热力学、动力学、均相和非均相催化剂、反应机理及反应工艺方面的研究进展。DMC和DEC酯交换是热力学上可自发进行的吸热反应。DMC和EtOH酯交换反应的热效应和自发性都较弱,符合二级动力学方程,反应速率与反应物浓度的平方成正比,可溶强碱催化时,生成EMC的活化能和指前因子较生成DEC的小;而可溶弱碱催化时,活化能和指前因子与强碱催化时相反。均相催化剂整体活性高,反应速率快,但易失活、可回收性差。文中以pKb值为标准,总结出可溶碱类催化剂的pKb值越低其碱强度越强,催化效率越高,针对工业采用的CH3ONa催化剂,描述了失活现象并提出失活机理。非均相催化剂重复使用性好,不存在后续分离问题,但是活性偏低、反应速率慢,金属氧化物类是现在研究最多的催化剂,其活性主要受催化剂酸碱性强弱及表面活性位数量的影响。碱催化酯交换是典型的亲核加成-消去反应,关键是烷氧基负离子的生成。催化精馏工艺中以CH3ONa/C2H5ONa为催化剂,塔顶采出DMC和MeOH共沸物,塔底三种酯类混合物经精馏分离得到目标产品,但均相催化剂易溶于产物,有少量残留就会造成EMC纯度下降;而非均相催化剂效率低、反应温度高,目前,相关工艺的研究报道极少。
尽管全球的固态锂电池研发和投资热闹非凡,但整体来看,固态锂电池尚处于研发阶段,固态锂电池存在界面阻抗大、快充难度大、成本高等一系列问题。在未来很长一段时间内(10-15年)使用液态电解液的锂电池仍处于市场主导地位。EMC具有锂离子溶解度高、稳定性好、凝固点低等一系列优点,作为锂电池电解液溶剂的主体地位不可替代。在未来锂电池级EMC的研究中,不仅要从提升产品纯度和降低生产成本的角度出发,而且要考虑减少能耗及绿色环保等问题,高效固体碱催化剂和涉及气、液、固三相的催化精馏技术是今后开发的重点和发展方向。
