团簇Co3FeP催化活性位点的研究
2021-09-08毛智龙方志刚井润田侯欠欠
毛智龙,方志刚,井润田,许 友,侯欠欠
(辽宁科技大学化学工程学院,辽宁鞍山 114051)
近年,非晶态合金由于具有短程有序、长程无序的特点,逐渐成为热点研究对象。向过渡金属Co 和Fe中加入类金属元素P 的非晶态合金Co-Fe-P 显示出优异的特性,如Co-Fe-P 体系磁性材料因具有三层空心微球和纳米线阵列等结构,在磁性材料、微波吸收材料和锂硫电池的介质等领域具有巨大的应用前景。Co-Fe-P体系催化剂存在多孔中空结构,此类结构有助于催化剂暴露出催化活性位点,促使其与反应物充分接触。Co-Fe-P 体系催化剂可催化水解析氧和水解析氢,也可整体催化水解;Co-Fe-P 非晶态合金也可与多种物质掺杂形成高效催化剂,如与MOF 衍生的二维掺氮碳纳米片耦合形成催化剂,将其封装入N,P 中一起掺杂至石墨烯形成催化剂等。综上所述,Co-Fe-P 体系非晶态合金不仅在材料与介质领域的应用值得期待,在作为催化剂的应用中也非常有前景。但目前学者多是从宏观层面对Co-Fe-P 体系催化剂进行研究,较少对其作用机理进行研究。鉴于此,以非晶态合金团簇CoFeP为局域结构模型,探讨其催化活性的影响机理,为下一步相关催化研究提供理论支撑。
1 团簇模型和计算方法
以团簇CoFeP为局域结构,利用密度泛函理论和拓扑学原理,在量子化学中较高的B3LYP/Lan12dz水平下,对团簇CoFeP 三重态和单重态的所有初始构型进行全参数的优化和频率计算。其中对Co,Fe原子采用18-eECP双ξ基组(3s,3p,3d/2s,2p,2d),对P原子采用Dunning/Huzinaga双ξ基组(9s,5p/3s,2p)并加上极化函数ξ
=0.55。所有的计算均在微型计算机hp Z440上用Gaussian09程序完成。2 结果与讨论
2.1 团簇Co3FeP的优化构型
经优化计算和排除相同构型,得到4 种单重态构型和5 种三重态构型,如图1。以能量最低的构型1为基准点(0 kJ/mol),其余构型按照相对能量大小依次排序,上标表示构型的自旋多重度。团簇CoFeP 的几何构型有平面四边形(构型5,0 kJ/mol),戴帽三角锥型(构型3,99.778 kJ/mol;构型4,786.279 kJ/mol),三角双锥型(构型1,626.355 kJ/mol;构型2,19.894 kJ/mol;构型4,131.920 kJ/mol;构型1,626.355 kJ/mol;构型2,626.752 kJ/mol;构型3,678.362 kJ/mol)。
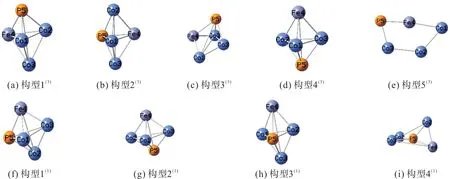
图1 团簇Co3FeP的优化构型Fig.1 Optimized configurations of cluster Co3FeP
2.2 团簇Co3FeP的催化活性
2.2.1 团簇CoFeP中不同原子对HOMO,LUMO轨道的贡献率
上世纪50 年代,日本理论化学家福井谦一提出了前线轨道理论,认为分子轨道中能量最高的占据轨道HOMO和能量最低的未占据轨道LUMO是决定化学反应发生的关键。因此,了解团簇CoFeP中各原子对HOMO和LUMO轨道的贡献率对确定团簇的催化活性位点很有必要。
表1 为团簇CoFeP 中不同原子对HOMO,LUMO 的贡献率。由表1 可看出:Co 原子对HOMO 和LUMO轨道的贡献率平均值超过50%,说明Co 原子可能是团簇CoFeP 的催化活性位点;单重态构型中,Co 原子对HOMO和LUMO轨道的贡献率超过50%,除构型2外的其他单重态构型中,Co原子对LUMO轨道的贡献率超过90%;三重态构型中,Co 原子对HOMO 和LUMO 轨道的贡献率只有部分超过50%,构型4和5中,Co原子对HOMO和LUMO轨道的贡献率未超过50%,说明Co原子对HOMO和LUMO轨道的贡献率受构型自旋多重度的影响。

表1 团簇Co3FeP中不同原子对HOMO,LUMO轨道的贡献率Tab.1 Contribution rate of different atoms in cluster Co3FeP to HOMO and LUMO orbitals
单重态构型中,Fe 原子对HOMO 和LUMO 轨道平均贡献率分别为37.366%和6.545%,P 原子只有5.071%和4.355%;除构型4外,Fe 原子对LUMO 轨道的贡献率比P 原子大,表明单重态构型中Fe 原子可能是团簇CoFeP 的另一个活性位点,说明单重态构型中团簇CoFeP 极有可能有Co,Fe 双催化潜在活性位点。三重态构型中,Fe 原子对HOMO 和LUMO 轨道的平均贡献率分别为33.122%和23.415%,P 原子为16.811%和25.548%。由此表明,Fe 原子对HOMO 和LUMO 的贡献率并没有整体上超过P 原子,在部分构型的贡献率中要小于P 原子。Co 原子在三重态构型中对HOMO 和LUMO 轨道的平均贡献率分别为50.014%和51.037%,说明三重态构型中团簇CoFeP可能是Co单催化潜在活性位点。
大部分构型中,P 原子对HOMO 和LUMO 轨道的贡献率较低,说明P 原子不太可能成为团簇CoFeP 的催化活性位点。构型5中P 原子的LUMO 轨道的贡献率为52.963%,产生了一个特殊现象。观察所有优化构型的几何形态可发现,只有构型5为平面构型,其他为立体构型,说明构型的几何形态影响构型中原子对HOMO和LUMO轨道的贡献率。
综上所述可知:单重态构型中可能含有Co,Fe 两个催化潜在活性位点,三重态构型中可能只有Co 一个催化潜在活性位点;构型中原子对HOMO 和LUMO 轨道的贡献率受其几何构型及构型自旋多重度的影响,说明构型的催化活性受其几何形态和自旋多重度的影响。
2.2.2 团簇CoFeP的催化能力
由前线轨道理论可得,一个化学反应的发生和HOMO,LUMO 轨道上的电子转移密切相关,能隙差越小,反应越易发生,催化活性越高。团簇CoFeP 的前线轨道能级HOMO(E
)、LUMO(E
)、费米能级(E
)和能隙差(E
)相关计算结果如表2。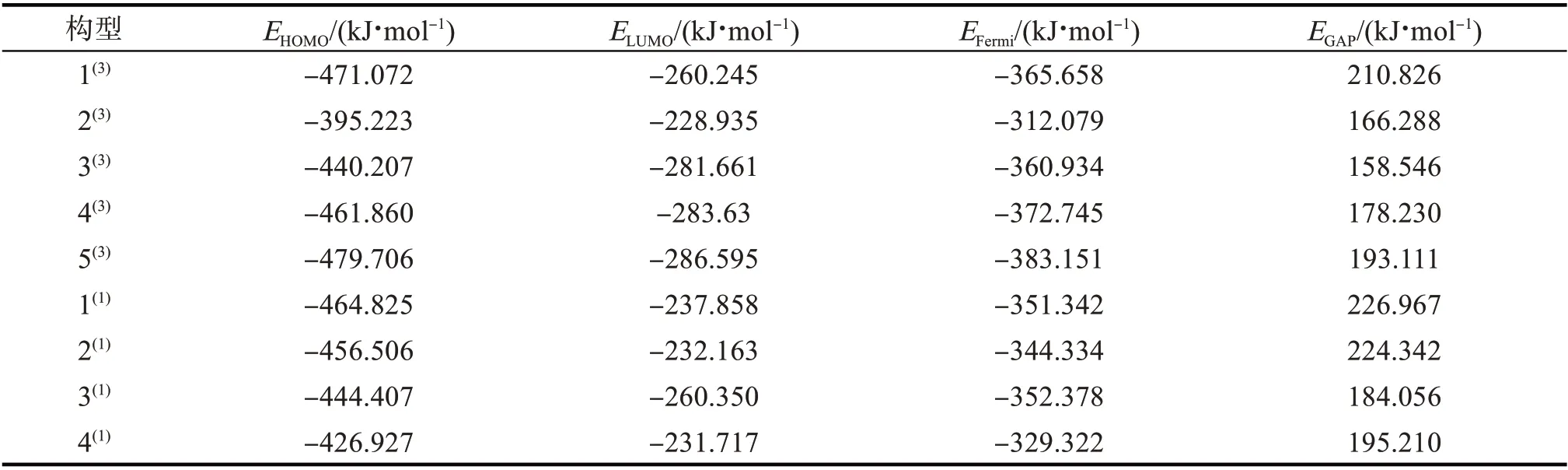
表2 团簇Co3FeP的前线轨道能级、能隙差及费米能级Tab.2 Front-line orbital energy level,energy gap difference and Fermi level of cluster Co3FeP
由表2可得:所有优化构型中,能隙差由大到小排列的构型为1,2,1,4,5,3,4,2,3;构型3的催化活性最高,构型1的催化活性最低;从构型的自旋多重度来看,三重态构型的催化活性普遍大于单重态。因此推断构型的催化活性受构型自旋多重度的影响。
为进一步确定团簇CoFeP 的催化活性是否受构型自旋多重度的影响,用费米能级进一步研究团簇CoFeP 的催化能力。团簇CoFeP 各优化构型的态密度(density of states,DOS)如图2,其费米能级态密度图附近最高峰的峰值及其与费米能级的距离如表3。图2 中:E
为费米能级左侧最高峰的峰值;D
为左侧最高峰与费米能级的距离;E
为费米能级右侧最高峰的峰值;D
为右侧最高峰与费米能级的距离。其中Fermi 能级左右两侧的峰值与Fermi 能级的距离分别表示失去或得到电子的能力,峰值表示得失电子的数量,Fermi 能级附近的电子云密度越大,催化活性越高。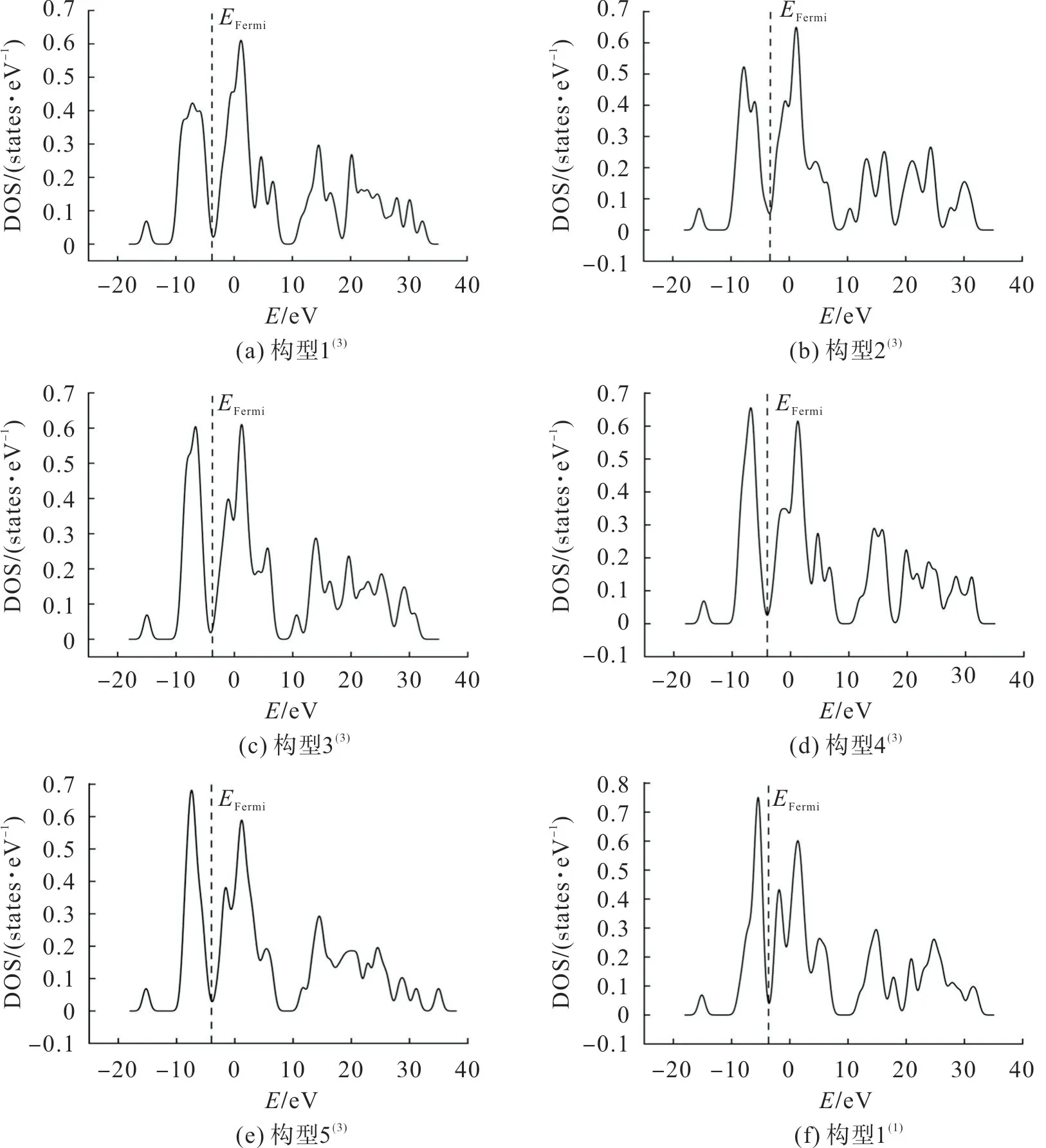
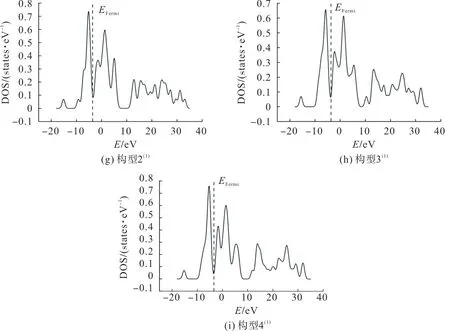
图2 团簇Co3FeP各优化构型的态密度Fig.2 DOS of each optimized configuration of cluster Co3FeP
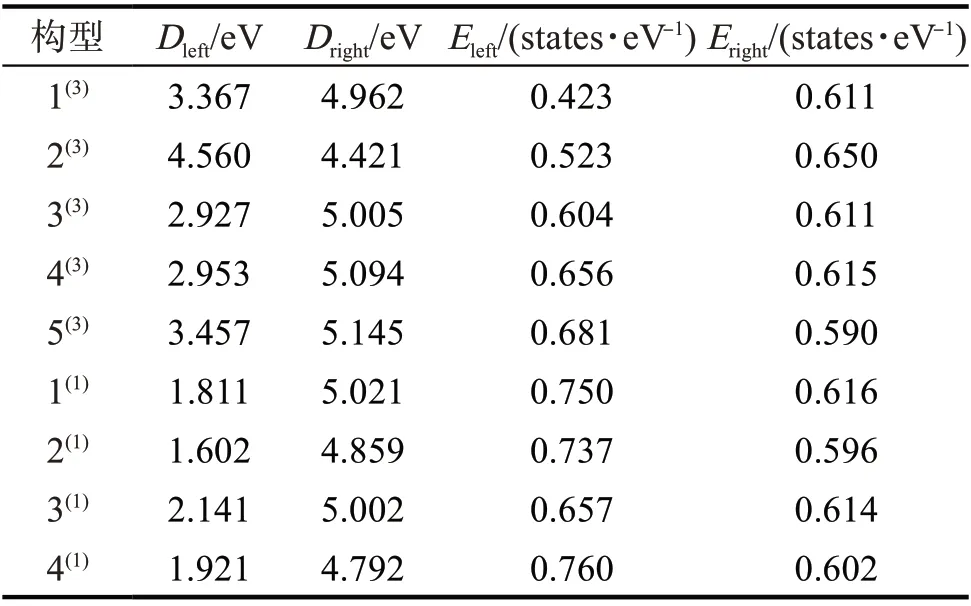
表3 团簇Co3FeP各优化构型中态密度图附近最高峰峰值及其与费米能级的距离Tab.3 Peak value of the highest peak near DOS and distance from the Fermi level in each optimized configuration of cluster Co3FeP
由图2和表3可看出:除构型2外,其他构型中左侧最高峰均离Fermi能级较近,说明这些构型易失去电子;值得注意的是构型2中左右两侧最高峰离Fermi能级距离相近,左右两侧峰值相差较大,说明构型2得失电子的能力相近,但得失电子的数量不同;构型3中,其左侧最高峰离Fermi能级距离比右侧最高峰近,且左右两侧峰值相差较小,说明构型3失电子的能力较强,得失电子的数量相近。
结合表3 可得,单重态的D
和E
与三重态的D
和E
相近,但单重态的D
比三重态的D
小且单重态的E
比三重态的E
大,表明单重态的左侧最高峰离Fermi 能级更近且峰值更大。说明在催化反应中单重态构型更易失去电子,且失去电子的数量大于三重态构型,进一步说明单重态构型催化活性比三重态构型高;也验证了上述结论,即在单重态构型中有Co,Fe双催化活性位点,三重态构型中只有Co单催化潜在活性位点。但催化活性位点发挥催化作用时不受任何干扰,故单重态构型中有Co,Fe两个催化活性位点时,其催化活性一定比三重态Co单个催化活性位点的高。通过能隙差可发现三重态的催化活性比单重态高,结合表1中的数据分析可得,Co对HOMO和LUMO轨道的平均贡献率超过50%,Fe在单重态构型中对HOMO和LUMO轨道的贡献率仅次于Co,但并没有发挥实际作用。若构型的自旋多重度等因素对其催化活性没有影响,则Co,Fe发挥作用时,单重态构型的催化活性应比三重态构型的高,且只有Co发挥作用单重态构型的催化活性应与三重态构型相近。但实际情况是三重态构型的催化活性比单重态的高。结合自旋多重度的定义可得,单重态构型没有成单电子,三重态构型有两个未成对电子,表明三重态构型更易得失电子。故在只有Co 原子发挥作用的情况下,三重态构型的催化活性不会与单重态相近,而是比单重态的高,与表2 中能隙差的情况相同。说明在团簇CoFeP 中只有Co 是有效催化活性位点,Fe 不是有效催化活性位点。由表3 还可得,除构型1,2和3外,其余构型为左侧最高峰大于右侧且左侧峰离Fermi 能级更近,说明在催化反应中这些构型失去电子数大于得到电子数,在催化反应中易催化亲电试剂的反应底物。综上可得:构型的自旋多重度影响构型的催化活性位点,进而影响其催化活性;单重态构型失电子能力比三重态强;所有构型中只有Co 为团簇CoFeP 有效催化活性位点;除构型1,2和3外,其余构型易催化反应底物中含有亲电试剂的反应。
2.2.3 团簇CoFeP的催化活性位点
引入库普曼斯定理对团簇CoFeP 的催化活性及催化活性位点进行研究。由前线轨道理论与库普曼斯定理可计算出团簇CoFeP 各优化构型的电离势E
、电子亲和能E
、电负性χ
和亲电指数ω
,结果如图3。其中:电离势表示失去电子需要的能量;电子亲和能表示得到电子需要的能量;电负性表示吸引电子的能力;亲电指数表示得到电子的能力,并且反应活性与电子亲和能、电负性和亲电指数成正相关,与电离势成反比。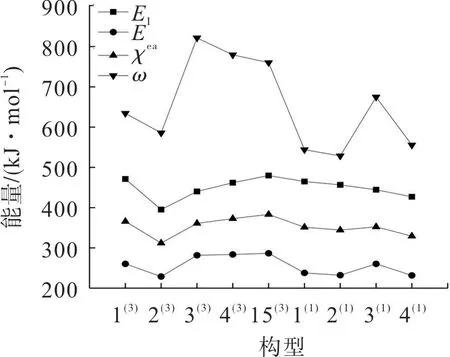
图3 团簇Co3FeP的电离势、电子亲和能、电负性和亲电指数Fig.3 Ionization potential,electron affinity,electronegativity and electrophilic index of cluster Co3FeP
由图3 可得:构型3,4和5的亲电指数、电子亲和能和电负性较大,说明上述构型得电子能力较强,较易生成阴离子,反应活性较大;构型4和5的电离势较大,说明构型4和5较难失去电子,构型4和5的催化活性不是最高的;构型3的电离势较小,说明型3的催化活性最高;三重态构型整体的电子亲和能、电负性和亲电指数比单重态大,三重态构型的电离势与单重态构型的电离势相近。表明三重态构型的催化活性比单重态高,与能隙差中的分析结论相似,可验证团簇CoFeP 催化性质中的结论。只有Co作用时单重态应与三重态的催化活性相近,但单重态的催化活性比三重态的催化活性低,说明催化活性位点发挥作用受构型自旋多重度的影响。
综上所述,构型3的催化活性最好,三重态和单重态的催化活性反常,进一步验证了Co 是团簇CoFeP唯一的有效催化活性位点。在判断构型的催化活性时不仅要考虑构型中催化活性位点的数量,也要考虑实际发挥作用的催化活性位点,还要考虑构型自旋多重度对活性位点发挥作用的影响。
3 结 论
以团簇CoFeP为局域结构模型,在较高的量子化学水平下,利用密度泛函理论、拓扑学原理和前线轨道理论及库普曼斯定理研究团簇CoFeP的催化活性,得出以下主要结论:
1)构型的几何形态和自旋多重度对团簇CoFeP 催化活性产生影响,在所有优化构型中,构型3的催化活性最好;
2)Co是团簇CoFeP中唯一的有效催化活性位点,Fe原子在实际得失电子中并未发挥作用,Fe不是有效催化活性位点;
3)除构型1,2和3外,其余构型易催化底物中含有亲电试剂的反应。
