无明显心脏病变的Emery-Dreifuss肌营养不良临床特征和基因分析
2018-11-07田秀秀袁宝玉吴迪钱方媛郭怡菁
田秀秀 袁宝玉 吴迪 钱方媛 郭怡菁
Emery-Dreifuss肌营养不良 (Emery-Dreifuss muscular dystrophy,EDMD)是一种罕见的遗传性疾病,儿童早期起病,主要临床表现为关节挛缩、进行性的肌无力和心脏受累。诊断主要依据临床表现与基因检测。EDMD患者无明显心肌受累与心电传导障碍者少见,现报告1例我科收治的Emery-Dreifuss肌营养不良2型无心脏受累病例。
1 临床资料
1.1 发病情况患者,女,14岁,因行走易摔倒12年,低头受限4年于2016年7月8日入我院。患者12年前走路易摔倒,起跳动作欠佳,后出现跟腱挛缩逐渐发展至不能起跳,9年前和7年前病情加重,于其他医院分别行“跟腱松解术”,术后症状缓解。4年前逐渐出现低头不能,持续无好转。自患病以来无明显肌肉萎缩、麻木及无力,无复视,无吞咽困难、饮水呛咳,无明显心悸胸闷。今发现仍存在跟腱挛缩 ,于我科就诊。自小饮食正常,睡眠正常,二便正常,无明显体重波动。患者为足月顺产,孕期胎心胎动正常,羊水无异常。出生后不能抬头,6个月左右会坐,9个月左右会爬,14个月时会站,18个月时会独立行走。自幼学习成绩中等,体育成绩略差,跑步较同龄儿慢,800米跑步无明显不适,跳远困难。患者父母体健,非近亲结婚。有一姐姐,体健,行动正常。家族中无类似病史。
1.2 体格检查神志清,精神正常。步入病房(正常步态)。面容狭长,高腭弓,查体合作,回答切题。双侧转颈、耸肩肌力5级,伸颈肌力5级,屈颈受限。 全身肌容正常,肌张力轻度增高,四肢肌力5级,双侧跟腱松解术后,跟腱轻度挛缩,双肘关节不能完全伸直,四肢腱反射(+),病理征(-)。骨盆压迫无疼痛,指地距40 cm,四字征(+)。心、肺、腹查体未见异常。
1.3 辅助检查心肌酶谱:肌酸激酶1672 IU/L(24~229 IU/L)。腰椎正侧位平片提示:腰椎生理曲度消失,脊柱轻度侧弯。骶髂关节CT未见明显异常。HLA B27(-)。超声心动图:静息状态下正常超声心动图表现。心电图:窦性心律,正常心电图。24 h动态心电图:正常。
神经肌肉电生理:NCV:右侧正中神经、双侧尺神经感觉运动传导速度、CMAP、SNAP波幅正常范围,F波出现率及潜伏期正常。EMG:右侧肱二头肌、三角肌、股四头肌、脊旁肌(L6、T9、L4)、腹直肌安静时未见自发电位,小力收缩运动单位电位时限正常,大力收缩募集正常。右侧拇短展肌安静时可见自发电位,小力收缩运动单位时限正常,大力收缩募集正常。短时及长时运动诱发实验无CMAP波幅下降。未见肯定肌源性或神经源性损害,未见强直性放电,运动诱发实验阴性。结论:①未见肯定肌源性或神经源性损害;②未见强直性放电。
肌肉磁共振:大腿股二头肌长头、腓内肌和比目鱼肌脂肪浸润明显,大收肌、半膜肌和腓外肌轻度脂肪浸润(见图1)。
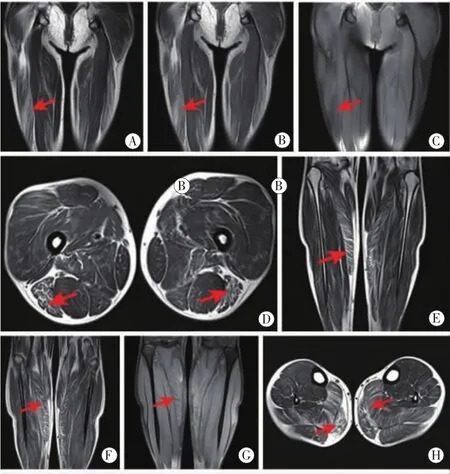
图 1 大腿 MRI冠状面 T1WI(A) 、T2WI(B)、横断面 T2WI(D)成像序列示:双侧大腿股二头肌长头内可见条纹状T2、T1高信号(红色箭头所示),冠状面PDWI压脂(C)呈稍低信号(红色箭头所示)。小腿 MRI冠状面 T1WI(E) 、T2WI(F)、横断面 T2WI(H)成像序列示:双侧小腿腓肠肌内侧头可见条纹状T2、T1高信号(红色箭头所示),冠状面PDWI压脂(G)呈稍低信号(红色箭头所示)
肌肉组织病理学(图2):肌纤维直径范围在15至100微米,可见散在变性、坏死肌纤维。部分肌纤维细胞核数目增加,可见中心核现象。肌纤维间结缔组织和血管未见显著异常。肌纤维中细胞色素C氧化酶 (COX)、NADH酶、琥珀酸脱氢酶(SDH)可见肌纤维染色局灶性减低,多和Ⅱ型纤维增多有关。单胺氧化酶(AMP)染色酶活性未见显著异常。腺苷三磷酸环化酶(ATPase)染色中,Ⅱ型肌纤维占优势。酸性磷酸酶活性在萎缩肌纤维中轻度升高。肌纤维糖原成分和脂肪成分大致正常。
基因学:本例选择了“肌肉疾病”的检测包(送检欧蒙医学诊断公司),包含了遗传性肌病、遗传性肌营养不良等,用高通量二代测序检测,发现LMNA 基因第7个外显子c.1366A>G疑似致病基因突变位点。针对这个位点,用Sanger法(一代测序,图3)进行验证。针对该位点,对其双亲进行了家系验证,结果显示为该基因位点的杂合突变。
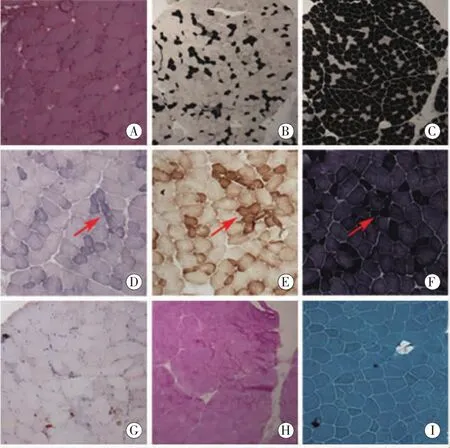
图2 A为HE染色,BC为腺苷酸环化酶(ATPase)染色,DEF分别为SDH、COX、NADH染色,G为油红染色,H为糖原染色,I为MGT染色

图3 患者及其双亲基因检测。A示患者带有c.1366A>G杂合突变;B、C示患者双亲基因未见相同位点突变
1.4 诊断、治疗与随访患者青少年女性,儿童早期发病,因行走易摔倒、低头受限入院。早期出现脊柱强直、关节挛缩,但无明显心肌受累与心电传导障碍。患者无晨僵及活动后脊柱强直症状缓解,且腰椎及骶髂关节影像学结果和HLA B27(-),可排除强制性脊柱炎诊断。基因检测发现LMNA基因新发突变,考虑LMNA基因相关肌病。结合患者临床表现为关节挛缩,脊柱强直,考虑诊断为EDMD2。目前该病尚无根治方法,最重要的是检测患者心脏受累情况,及时治疗。建议患者25岁之前,每年行心电图、超声心电图和24 h动态心电图进行监测。25岁之后,每5年监测一次。
2 讨论
本例患者基因学检测结果为LMNA基因的一个疑似致病突变c.1366A>G,p.Asn456Asp (在基因 检 测 中 覆 盖 了 ACTA1,ANO5,BAG3,CAPN3,CAV3等,均未发现其他相关疾病基因的突变),针对该位点,对其双亲进行了家系验证,结果显示该患者为该基因位点的杂合突变。家族中无类似病史,患者父母均未携带此基因,结合患者早期出现的关节挛缩与脊柱强直,疑似EDMD2型常染色体显性遗传。该患者为LMNA基因新发突变(novel mutation),该突变位点为国内首次报道,目前仅见1例国外报道[1]。
Emery-Dreifuss肌营养不良是一种罕见的相对良性的遗传性疾病 。1966年首先由Emery和Dreifuss描述,Rowland于1979年提议将该病命名为EDMD。关节挛缩、进行性的肌无力和心脏受累是该病的三大临床特征。通常在儿童早期发病,病程的前10年以关节挛缩和脊柱强直常见,30岁左右出现肌肉萎缩与无力,常呈肱-腓分布,逐渐进展至肩胛骨和骨盆带肌肉。心脏受累相对出现较晚,包括心律失常、扩张型心肌病、心力衰竭、猝死等。EDMD按照遗传方式不同,分为X连锁隐性遗传EDMD(X-linked recessive inheritance,XLEDMD)、常染色体显性遗传EDMD(autosomal dominant inheritance,AD-EDMD)和常染色体隐性遗传EDMD (autosomal recessive inheritance,AR-EDMD)。XL-EDMD的和AD-EDMD的临床表现相似,通常在儿童早期出现关节挛缩,亦存在不同之处。XL-EDMD患者最早出现关节挛缩,而ADEDMD通常在肌无力后出现关节挛缩。XL-EDMD肌肉萎缩和无力进展缓慢,30岁时病情加重,ADEDMD肌肉萎缩和无力变化与年龄无明显相关。AD-EDMD心脏受累多表现为左心室扩张和功能障碍的室性快速性心律失常和扩张型心肌病。AR-EDMD与AD-EDMD的主要致病基因均为LMNA基因,但 AR-EDMD通常临床症状较重,早期出现严重的肌营养不良和关节挛缩。
AD-EDMD多见于LMNA基因头和尾区的错义突变,Lamin A/C突变主要引起与横纹肌、脂肪组织周围神经和多系统早衰等相关疾病,最常见的是骨骼肌和心肌受累[2]。LMNA基因突变相关的肌病多有心脏受累[3]。本例患者临床表现与典型的EDMD不同,主要表现为关节挛缩与脊柱强直,尚无心脏受累临床表现,这一特征可能为该突变位点特有的临床表现。 SUSANA等[1]收集了15例LMNA基因新发突变患者,其中一名患者的突变位点与本例患者相同,初始表现为关节挛缩,逐渐出现颈部及近端肌肉无力,同样无心肌受累表现。最新的一篇关于EDMD的基因综述表明EDMD出现心脏受累一般在发病20年以上,表现为心悸、晕厥、运动不耐受和充血性心力衰竭[4]。本例患者病程不足20年,需随访心脏受累情况。
本例患者发育迟缓,早期出现脊柱强直,需与以下疾病鉴别。强制脊柱肌营养不良(rigid spine muscular dystrophy1,RSMD1)、Ullrich 型先天性肌营养不良 (Ullrich congenital muscular dystrophy,UCMD)、先天性肌营养不良(congenitalmuscular dystrophy type 1A,MDC1A)三者均发病较早,运动发育迟滞,并出现脊柱强直。RSMD1主要临床表现为肌强直,进行性肌无力,特征性斧头脸。UCMD往往伴发特有的近端关节挛缩(肩关节、肘关、膝关节)合并远端关节弹性过度,肌无力症状较重。 MDC1A患者头颅MR可见白质病变,部分患者可出现智能发育迟缓或癫痫等中枢神经系统受累症。
本例影像学表现以大腿股二头肌长头、腓内肌和比目鱼肌脂肪浸润明显,大收肌、半膜肌和腓外肌轻度脂肪浸润。有学者收集了10例EMD与32例LMNA基因突变的患者,两种致病基因的患者MRI具有相同的脂肪浸润模式,主要累及的肌肉为椎旁、臀肌、股四头肌、股二头肌、半腱肌、半膜肌、大收肌(adductor major)、比目鱼肌和腓肠肌[5]。MERCURI等[6]认为 EDMD2型患者肌肉 MRI,在大腿层面,所有股肌都有可能出现不同程度的选择性受累;在小腿层面,更易出现腓肠肌内侧头受累,比目鱼肌也常受累。本例患者表现为部分下肢肌肉的不同程度的选择性受累。病理学改变为散在变性、坏死肌纤维,部分肌纤维细胞核数目增加,少量中心核,Ⅱ型肌纤维优势。有研究发现EDMD患者肌肉组织病理学表现为非特异性肌病或营养不良改变,包括纤维大小的变化,内核的增加,肌内结缔组织的增加和肌纤维坏死[4]。肌肉活组织检查观察到的肌肉营养不良的变化缺乏特异性。肌电图通常表现为正常神经传导的肌病特征[4],本例肌电图结果亦为阴性。部分EDMD 2患者肌电图可见大量MUAPs波幅增高[7],亦可出现肌源性损害[8]。
本例患者早期出现的关节挛缩与脊柱强直,以踝关节和肘关节为著,暂无明显肌肉萎缩、无力,亦无明显心肌受累与心电传导障碍。双亲基因型正常,患者为LMNA基因散发杂合突变,c.1366A>G,p.Asn456Asp突变位点的在LANA基因较为罕见。无明显心脏病变可能为该新发突变特有的临床表现。
