具有氨基和羧基间单氢键的α-Ala分子旋光异构机理及水和羟自由基的作用
2018-09-12田子德杨晓翠王佐成
田子德,高 峰,杨晓翠,王佐成
(1.白城师范学院 理论计算研究中心,白城 137000; 2.白城师范学院 物理学院,白城 137000; 3.白城师范学院 数学学院,白城 137000)
α-丙氨酸(α-Ala)是组成人体蛋白质的重要氨基酸之一,根据其构象和旋光性的不同分为左旋体(S-α-Ala)和右旋体(R-α-Ala),两种手性对映体具有不同的功能特性.S-α-Ala有预防肾结石、协助葡萄糖代谢、缓和低血糖等作用.R-α-Ala有抑菌作用,是保湿因子的主要成分,用于手性药物和手性助剂等领域.已有实验研究[1-2]证实生命体内存在微量的R-α-Ala,体内过量的R-α-Ala可导致某些疾病和过早衰老,并猜测他的部分来源是左旋体的异构化.
由于α-Ala的重要性以及体内有微量R-α-Ala存在,人们对α-Ala进行了广泛的研究.Stepanian和Adamowicz组[3-4]用密度泛函理论的B3LYP方法,对α-Ala两种异构体进行了理论研究,得到了与实验数据较吻合的α-Ala分子构象.王文清组[5]利用单晶中子衍射研究了295K和60K时,α-Ala手性对映体的结构特征以及由S-α-Ala向R-α-Ala转变的可能性.文献[6-8]的研究表明: 以羰基、羧基以及羧基和甲基联合作质子迁移桥梁,α-Ala分子手性转变裸反应决速步的能垒分别为326.6kJ·mol-1、316.3kJ·mol-1和337.4kJ·mol-1;在水环境下,以羰基作为氢迁移桥梁,手性转变反应决速步的能垒被降到173.0kJ·mol-1;羧基内先氢迁移,然后手性碳上的氢再以羰基为桥梁迁移,手性转变反应决速步的能垒被降到167.8kJ·mol-1.文献[9]的研究表明: 随着扶手椅型SWBNNT孔径尺寸的减小,α-Ala手性转变决速步能垒急剧减小,在SWBNNT(5,5)内决速步吉布斯自由能垒为48.1Kcal·mol-1(201.1kJ·mol-1),是质子从手性碳向氨基氮和质子从氨基氮向羰基氧双质子协同迁移的过渡态产生的.
虽然人们对α-Ala分子的结构特性及手性转变机理的研究已较深入,研究结果对解释生命体内α-Ala的旋光异构以及在实验上实现α-Ala的旋光异构有一定的积极作用.但167.8kJ·mol-1已经达到了质子迁移的“极限能垒”167.0kJ·mol-1[10-11],纵使考虑到生命体内温度的涨落、分子的碰撞,被越过的几率也是很小,还要考虑某种异构酶的催化作用.这些研究结果对于解释生命体内α-Ala的旋光异构,不是十分理想.201.1kJ·mol-1已经远高于质子迁移的“极限能垒”167.0kJ·mol-1[10-11],SWBNNT(5,5)还不能作为实现α-Ala旋光异构的理想的纳米反应器.因此,有必要对α-Ala的旋光异构进行深入的研究.
大气富含水汽,水是重要的溶剂,生命体是富水环境并有少量的对生物体危害极大的羟基自由基存在[12].已有文献[13-14]研究表明,氨基作质子迁移桥梁的反应通道是氨基酸分子实现手性转变反应的优势通道,文献[7-8,14-15]研究表明,水分子和水分子团簇对质子迁移过程具有催化作用.基于此,本工作研究了氨基作氢迁移桥梁α-Ala分子的旋光异构、水分子簇和羟自由基的作用及水溶剂效应.
1 研究与计算方法
采用密度泛函理论的B3LYP[17-18]方法,在B3LYP/6-31+G(d,p)水平,全优化反应过程的稳定点和过渡态[19-20].为计算高水平的反应过程势能面,采用微扰理论的MP2方法[21-22],在MP2/6-311++G(2df,pd)水平计算单点能,利用Etotal=ESP+EZPV(ESP为高水平的单点能,EZPV为零点振动能,Etotal为高水平的总能量)计算总能量.对H迁移过程,把水视为离散介质,水分子直接参与反应.溶剂效应的计算,采用气相的构象,利用自洽反应场(SCRF)理论的smd模型方法[23]计算水连续介质的溶剂化单点能.α-Ala 分子与2个水分子通过氢键作用形成的络合物分子记为α-Ala·2H2O,水溶剂环境下记为α-Ala·2H2O@water,其余体系的表示法类似.通过对过渡态虚频振动模式的分析和内禀反应坐标(IRC)[24-25]计算,对过渡态进行确认.所有计算均由Gaussian09[26]软件完成.
2 结果与讨论
2.1 气相α-Ala分子的手性转变机理
文献[27-28]研究表明,氨基酸的羟基与氨基之间为分子内单氢键时氨基酸分子构型最稳定.基于此,对羟基与氨基之间为分子内单氢键的S-α-Ala和R-α-Ala的几何构型进行优化,得到图1所示的互为对映体的结构.

图1 单体S与R型α-Ala的结构Fig.1 The structure of monomer S-type α-Ala and R-type α-Ala
研究发现: 氨基作H迁移桥梁,实现α-Ala由S型向R型的异构可有两个反应通道.一是分步机理,羧基实现从反式平面结构向顺势平面结构的异构后,手性碳上的质子再以氨基为桥从纸面外迁移到纸面里,命名为a通道;二是协同机理,S-α-Ala先经过质子从手性碳向氨基氮迁移和羧基从反式平面结构向顺势平面结构异构协同进行的过渡态,异构成第1中间体.然后,质子化氨基上的一个质子再在纸面里从氨基氮迁移到手性碳,命名为b通道.下面对S-α-Ala在这两个通道的手性转变机理分别进行讨论.
S-α-Ala在a通道上的反应历程见图2(a),首先S-α-Ala经过羧基上的12H绕9C-10O键轴旋转的过渡态a_S-TS1,异构成中间体a_S-INT1.二面角12H-10O-9C-11O从179.12°变为1.50°,实现了羧基从反式向顺式平面结构的异构.结构分析表明,从S-α-Ala到过渡态a_S-TS1过程骨架结构基本没变,只是羟基旋转,二面角12H-10O-9C-11O从179.12°变为90.96°.因此,a_S-TS1产生的能垒不高,只有48.41kJ·mol-1(图3,见第520页).然后,中间体a_S-INT1_1经过渡态a_TS2_1,实现12H从手性碳1C向氨基氮6N的迁移,异构成第2中间体a_INT2.从a_S-INT1到a_TS2,1C-13H键从0.10937nm拉伸到0.13810nm断裂;1C-6N键从0.14644nm拉伸到0.15873nm断裂;骨架二面角6N-1C-4C-9C从124.49°变为141.20°,骨架形变较大.此过程两个化学键断裂并且骨架形变较大,a_TS2会产生较高的能量,能垒值是267.41kJ·mol-1.接着a_INT2经过渡态a_TS3,实现7H在纸面里从氨基氮6N向手性碳1C的迁移,异构成中间体a_R-INT3,实现手性转变.从a_INT2到a_TS3,6N-7H键长从0.10322nm拉伸到0.11636nm断裂;1C-6N键长从0.14859nm拉伸到0.15873nm断裂.此过程的键长伸长小于从a_S-INT1到a_TS2过程,并且骨架二面角6N-1C-4C-9C从163.78°变为-141.20°,α-碳1C从sp2杂化向sp3杂化过渡,是放热过程.因此a_TS3_1产生的能垒会远远小于a_TS2产生的能垒,只有106.46kJ·mol-1.然后,a_R-INT3经过氨基上的13H和8H左右翻转的过渡态a_R-TS4,异构成a_R-INT4.结构分析表明,此过程骨架结构基本没变,只是二面角13H-6N-1C-8H从-119.88°变为119.95°,非骨架原子翻转异构只需较少的能量,因此,a_R-TS4产生的能垒不高,只有17.20kJ·mol-1.最后,a_R-INT4经12H绕9C-10O键轴旋转的过渡态a_R-TS5,异构成产物a_P_R-α-Ala.a_P_R-α-Ala分子的5N-12H距离是0.18951nm,其具有氨基和羧基之间较强的单氢键作用,结构稳定.此基元反应相似于第1基元反应,a_R-TS5产生的能垒不高,只有43.22kJ·mol-1.由与a_R-TS4和a_R-TS5的双向势垒都较低,常温下容易越过,因此,S-α-Ala在a通道旋光异构的产物是a_R-INT3、a_R-INT4和a_P_R-α-Ala共存,只是a_P_R-α-Ala的含量较高.

图2 S-α-Ala的手性转变过程、驻点结构和过渡态的虚频振动模式Fig.2 The chiral transition process, stationary points structure and transition state imaginary frequency of S-α-Ala
S-α-Ala在b通道手性转变的第1基元反应,见图2(b),S-α-Ala经手性碳1C上的13H向氨基氮6N迁移与12H绕9C-10O键旋转协同进行的过渡态b_TS1,异构成中间体b_INT1,实现了氨基质子化,同时实现了羧基从反式向顺式平面结构的异构.结构分析表明,b_INT1的构象全同于a_INT2.其以后的异构过程同于a_INT2的异构过程,不再赘述.从S-α-Ala到b_TS1,1C-13H键长从0.10974nm增加到0.14200nm断裂;1C-6N键长从0.14768nm增加到0.16107nm键断裂;19H绕17O-15C旋转,二面角12H-10O-9C-11O从179.12°变为-162.20°;骨架二面角6N-1C-4C-9C从124.68°变为134.32°,骨架伸展形变较大.两个化学键断裂需要较大的能量,羟基的旋转和骨架的形变也需能量,并且S-α-Ala到b_TS1过程键长拉伸幅度大于a_S-INT1到a_TS2过程.因此,b_TS1产生的能垒较a_TS2高出许多,此基元反应的能垒为326.90kJ·mol-1(图3).

图3 α-Ala手性对映体转变反应过程的势能面示意图Fig.3 Potential energy surfaces diagram of chiral enantiomer transition reaction of α-Ala
从图3可以看出,反应通道a具有优势,为主反应通道,质子从手性碳向氨基迁移过程是决速步骤.决速步内禀能垒是267.41kJ·mol-1.
2.2 水分子簇的催化及水溶剂效应
文献[7-8,12-14]的研究表明,2个水分子簇对质子从手性碳向氨基氮迁移和质子从质子化氨基向手性碳迁移的过程有较好的催化作用.篇幅所限,仅讨论2个水分子簇对优势通道第2和3基元反应(质子迁移过程)的催化.考虑到水汽浓度较大时,产物中间体a_INT2·2H2O(m)(水分子可以在不同的方位与a_INT2络合,m和n分别表示2H2O在底物分子的前面和后面,见图4(a))不需脱水,就可以在2个水分子簇作用下实现质子在纸面里从氨基氮向手性碳迁移,异构成a_R-INT3·2H2O(m)·2H2O(n).还计算了a_INT2·2H2O(m)在2个水分子簇作用下,向a_R-INT3·2H2O(m)·2H2O(n)异构的过程.反应历程分别见图4(a)、(b)和(c),手性转变反应过程的势能面剖面分别见图5(a1)、(a2)和(b)(第522页).水溶剂环境下手性转变反应过程的势能面剖面图见图5中的@water曲线.
对第2基元反应,首先2个水分子簇在a_S-INT1的前面13H和6N的方位与a_S-INT1通过氢键作用形成前驱络合物a_S-INT1·2H2O(m),a_S-INT1·2H2O(m)的氢键能是58.89kJ·mol-1.然后,a_S-INT1·2H2O(m)经过渡态络合物a_TS2·2H2O(m)异构成中间体产物络合物a_INT2·2H2O(m).过渡态a_TS2·2H2O(m)的七元环结构中,氢键1C-13H-14O、14O-17H-18O和18O-15H-6N的键角分别为161.10°、160.37°和161.49°,趋于平角,这3个氢键较强;二面角1C-13H-14O-17H、14O-17H-18O-15H和17H-18O-15H-6N的键角分别为-24.59°、-8.49°和-35.13°,偏离平角较小,过渡态a_TS2·2H2O的七元环结构接近平面,张力小,结构比较稳定.又从a_S-INT1·2H2O(m)到a_TS2·2H2O(m)过程,骨架二面角6N-1C-4C-9C从128.18°变为128.30°,骨架没有发生形变,因此,a_TS2·2H2O(m)产生的能垒不会十分高.但是,从a_S-INT1·2H2O(m)到a_TS2·2H2O(m)过程,1C-13H、14O-17H和18O-15H键长从0.11001nm、0.09804nm和0.09932nm分别伸长到0.13314nm、0.14697nm和0.16130nm断裂;1C-6N从0.14763nm伸长到0.15160nm.因此,a_TS2·2H2O(m)产生的能垒又不会太低,此基元反应的内禀能垒是131.77kJ·mol-1.从图5(a1)还可以看出,溶剂效应使此基元反应的内禀能垒降到107.83kJ·mol-1.
对第3基元反应,首先是2个水分子簇在a_INT2的后面15H和1C的方位与a_INT2通过氢键作用形成前驱络合物a_INT2·2H2O(n),a_INT2·2H2O(n)的氢键能是98.56kJ·mol-1.然后,a_INT2·2H2O(n)经过渡态络合物a_TS3·2H2O(n)异构成中间体产物络合物a_R-INT3·2H2O(n).亦即,质子以2个水分子簇为媒介,在纸面里从a_INT2的氨基氮迁移到了手性碳,实现了手性转变.a_TS3·2H2O(n)和a_TS2·2H2O(m)相似,七元环过渡态比较稳定,不会产生十分高的能垒.又从a_INT2·2H2O(n)到a_TS3·2H2O(n)过程,1C-6N键长从0.15048nm拉伸到0.15160nm,键长6N-15H、18O-17H和13O-14H从0.09924nm、0.10436nm和0.10111nm分别拉伸到0.10716nm、0.10496nm和0.12949nm,拉伸幅度均比a_S-INT1·2H2O(m)到a_TS2·2H2O(m)小许多;骨架二面角6N-1C-4C-9C从-137.03°变为-128.31°,α-碳1C从sp2杂化向sp3杂化过渡,是放热过程.因此a_TS3·2H2O(n)产生的能垒会远远小于a_TS2·2H2O(m)产生的能垒,只有10.50kJ·mol-1.从图5(a2)可以看出,溶剂环境下此基元反应的内禀能垒是11.74kJ·mol-1,略微升高.
从图4(c)可以看出,水汽浓度较大时,中间体产物络合物a_INT2·2H2O(m)的a_INT2和H2O之间的分子间氢键不需断开,a_INT2·2H2O(m)可以与其右侧的2个H2O分子通过氢键作用,形成4水氢键络合物a_INT2·2H2O(m)·2H2O(n),a_INT2和4H2O(n)之间的氢键能是155.75kJ·mol-1(a_INT2·2H2O(m)和2H2O(n)之间的氢键能是155.75kJ·mol-1与98.56kJ·mol-1之差,是57.16kJ·mol-1),a_INT2·2H2O(m)·2H2O(n)经过渡态a_TS2·2H2O(m)·2H2O(n)异构成产物a_R-INT3·2H2O(m)·2H2O(n),S-α-Ala实现了手性转变.从势能面剖面图5(b)可知,此基元反应在气相的内禀能垒是26.08kJ·mol-1,在水溶剂相时降到18.74kJ·mol-1.
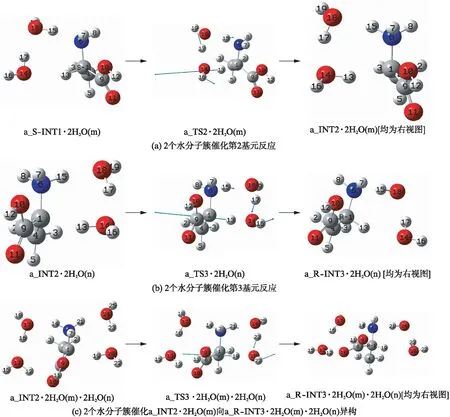
图4 水分子簇催化的质子迁移异构过程Fig.4 The proton migration isomerism processes catalyzed by water clusters
结合图5的(a1)和(a2)和图3可以看出,水溶剂化效应对羧基异构反应(第1和5基元反应)的能垒基本没有影响,使氨基左右翻转异构的能垒略有升高(第4基元反应),水分子簇的催化与水溶剂效应的共同作用,使决速步内禀能垒降到107.83kJ·mol-1,已经远低于质子迁移的“极限能垒”167.0kJ·mol-1[10-11],但还是远远高于温和反应的能垒84.01kJ·mol-1[10-11],这说明水环境下S-α-Ala可以缓慢地实现手性转变.
水汽相反应过程中,某一基元反应所放的热量可以被下步反应利用,总反应能垒要用表观能垒描述.从图5的(a1)和(b)可以看出,水汽浓度较大时,手性转变过程决速步的表观能垒是72.88kJ·mol-1,低于温和反应的能垒84.01kJ·mol-1[10-11],说明水汽浓度较大时,α-Ala的手性转变可以较快地实现.从图5(b)可以看出,第3基元反应是放热反应.

图5 水分子簇催化质子迁移过程及水溶剂环境下手性转变过程的势能面示意图Fig.5 Potential energy surfaces diagram of proton transfer process catalyzed by water clusters and chiral transition in water solvent environment
从图5(a1)可以看出,2水合物a_INT2·2H2O(m)的a_INT2和2个H2O分子的氢键断裂需克服的氢键能是98.56kJ·mol-1,因此,a_INT2·2H2O(m)的氢键很难断裂.从图5(a1)和(a2)可以看出,从a_S-INT1+2H2O到a_INT2+2H2O的表观能垒是160.94kJ·mol-1,说明水汽浓度较小时,α-Ala分子的手性转变很难实现.
2.3 羟自由基致丙氨酸损伤机理及水溶剂化效应
研究表明,第1基元反应同于2.1节的讨论,羟自由基不起作用.羟自由基对α-Ala异构的第2基元反应(氢迁移反应)起作用,并且是羟自由基与水分子以氢键结合成链共同起作用,导致a_S-INT1损伤,亦即羟自由基致α-Ala损伤.这又分两种情形,一种是水分子抽氢机理,另一种是羟自由基抽氢机理,下面分别进行讨论:
第1种情形,水分子抽氢致a_S-INT1损伤的反应历程见图6(a)(本节未对中间体反应物和产物与羟自由基及水分子之间氢键能进行讨论),反应过程的吉布斯自由能势能面见图7(a)(第524页).

图6 羟自由基水分子链致a_S-INT1损伤的反应历程Fig.6 Reaction process of a_S-INT1 damage induced by hydroxyl radical water molecules chains
首先,a_S-INT1通过与一个水分子和一个羟自由基通过氢键作用形成氢键络合物a_S-INT1·(H2O·OH·),然后a_S-INT1·(H2O·OH·)经过渡态a_TS2·(H2O·OH·)形成产物a_P1*·2H2O.a_P1*已经没有了手性,失去了α-Ala分子的特性,亦即α-Ala分子损伤,这里星号表示a_P1是氨基酸损伤的产物.
七元环结构过渡态a_TS2·(H2O·OH·)的氢键角1C-13H-17O、17O-15H-14O和14O-12H-5N分别是160.66°、158.62°和165.70°,二面角1C-13H-17O-15H、17O-15H-14O-12H和15H-14O-12H-5N分别是12.03°、-8.10°和2.81°,七元环结构基本共面,因此,过渡态a_TS2·(H2O·OH·)较稳定;从a_S-INT1·(H2O·OH·)到a_TS2·(H2O·OH·),反应活性中心骨架二面角5N-1C-3C-8C从129.01°变为128.69°,基本没变.因此,从a_S-INT1·(H2O·OH·)到a_TS2·(H2O·OH·)不需要太多的能量.但从a_S-INT1·(H2O·OH·)到a_TS2·(H2O·OH·)过程,1C-13H、17O-15H和14O-12H分别从0.10998nm、0.09716nm和0.10085nm拉伸到0.13837nm、0.14775nm和0.17313nm,并且断裂.3个化学键的拉伸断裂还是需要一定的能量的,因此,a_TS2·(H2O·OH·)产生了124.76kJ·mol-1的能垒.对a_TS2·(H2O·OH·)进行的IRC计算表明,从a_TS2·(H2O·OH·)到a_P1*·2H2O过程,首先,12H迁移到氨基氮5N,形成质子化氨基;然后,羟自由基14O-15H形成,同时羟自由基又拔掉5N上的12H;在12H向14O回迁的过程中,13H转移到17O;最后,12H迁移到14O与14O-15H形成水分子,丙氨酸损伤的产物与水分子的氢键络合物a_P1*·2H2O形成.
络合物a_P1*·2H2O分子的氢键距离13H-10O和15H-17O分别是0.17986nm和0.18205nm,氢键键角17O-13H-10O和14O-15H-17O分别是174.31°和171.54°,说明损伤产物a_P1*·2H2O的两个水分子之间以及水分子17O-13H-16H和a_P1*之间存在较强的分子间氢键.14O-2H的距离是0.23407nm,说明水分子14O-12H-15H和a_P1*之间存在范德华作用.a_P1*的骨架原子二面角5N-1C-3C-8C和3C-1C-8C-9O分别为-179.78°和179.06°,说明a_P1*的骨架原子之间形成了共轭的∏键.因此,a_P1*·2H2O的构型十分稳定.124.76kJ·mol-1的能垒已经低于质子迁移的“极限能垒”167.0kJ·mol-1[10-11],但还是远远高于温和反应的能垒84.01kJ·mol-1[10-11],这说明水汽环境下羟自由基的存在可导致S-α-Ala缓慢地损伤.

图7 羟自由基水分子链致a_S-INT1损伤反应过程的势能面Fig.7 Potential energy surfaces diagram of reaction process of a_S-INT1 damage induced by hydroxyl radical water molecule chains
从图7(a)可以看出,水溶剂化效应使损伤过程的能垒降到6.59kJ·mol-1,这远远低于温和反应的能垒84.01kJ·mol-1[10-11],这说明水溶剂环境下羟自由基的存在可导致S-α-Ala迅速地损伤,水溶剂起了极好的助催化作用.水溶剂化效应使羟自由基致α-Ala损伤过程的能垒大幅下降原因是: a_TS2·(H2O·OH·)的偶极矩是3.7721,远大于a_S-INT1·(H2O·OH·)的偶极矩1.9955,因此,在极性溶剂水中前者比后者更容易极化,a_TS2·(H2O·OH·)的能量降低会比a_S-INT1·(H2O·OH·)的能量降低显著,导致水溶剂环境下羟自由基致α-Ala损伤的能垒大幅下降.产物a_P1*·2H2O的偶极矩是2.6547,也比a_S-INT1·(H2O·OH·)的偶极矩大,极性水溶剂效应会使前者的能量相对后者降幅大些,因此,水溶剂环境下a_P1*·2H2O变得比a_S-INT1·(H2O·OH·)相对更稳定.
第2种情形,羟自由基抽氢致a_S-INT1损伤的反应历程见图6(b),反应过程的吉布斯自由能势能面见图7(b).首先,a_S-INT1与一个水分子和一个羟自由基通过氢键作用形成氢键络合物a_S-INT1·[(OH·)·H2O].然后,a_S-INT1·[(OH·)·H2O]经过渡态a_TS2·[(OH·)·H2O]实现了13H从手性碳向羟自由基的迁移,形成产物a_P2*·2H2O.a_P2*已经没有了手性,失去了α-Ala分子的特性,亦即α-Ala分子损伤.
从图6(b)可以看出,羟自由基抽氢致a_S-INT1损伤过程中,水分子15H-18H-17O并没参与反应,只是起到稳定羟自由基处于理想的利于抽氢位置的作用.从a_S-INT1·[(OH·)·H2O]到a_TS2·[(OH·)·H2O]过程,1C-13H从0.11001nm拉伸到0.11585nm,拉伸幅度仅有0.00584nm;反应活性中心骨架二面角6N-1C-4C-9C从128.01°变为131.74°,基本没变.一个化学键较小幅度的拉伸,需要的能量很小,因此,a_TS2·[(OH·)·H2O]产生的能垒较低,只有21.56kJ·mol-1.这能垒远低于温和反应的能垒84.01kJ·mol-1[10-11],这说明水汽环境下羟自由基拔氢可致S-α-Ala较快地损伤.结构分析表明,相似于a_P1*·2H2O,损伤产物a_P2*·2H2O的两个水分子之间以及水分子14O-13H-16H和a_P1*之间存在较强的分子间氢键,水分子17O-18H-15H和a_P1*之间存在范德华作用,a_P2*的骨架原子之间形成了共轭的∏键,因此,a_P2*·2H2O的构型十分稳定.
从图7(b)可以看出,水溶剂化效应使损伤过程的能垒微升到22.4kJ·mol-1,损伤反应过程仍为低势垒过程,这说明水溶剂环境下羟自由基拔氢致S-α-Ala也较快地损伤,水溶剂同样起了微弱的负催化作用.
从图7可看出,羟自由基水分子链联合致a_S-INT1损伤反应过程均为放热反应.无论是水汽相还是水溶剂相,在水分子拔氢和羟自由基拔氢两个通道的竞争中,水为汽相时羟自由基拔氢通道具有明显的优势,水为液相时水分子拔氢通道具有优势.
3 结 论
标题反应有两个通道a和b,a是羧基先异构,然后质子以氨基为桥迁移的分步机理;b是质子迁移与羧基异构同步进行的协同机理.a为优势通道,质子从手性碳向氨基迁移过程是决速步骤.2个水分子簇作氢迁移媒介,使决速步内禀能垒从裸反应的267.41kJ·mol-1降到131.77kJ·mol-1,水溶剂效应使该能垒降到107.83kJ·mol-1.羟自由基水分子链联合作用使α-Ala损伤,水分子拔氢和羟自由基拔氢的能垒分别是124.76和21.56kJ·mol-1,水溶剂效应使两个能垒降到6.59和-186.99kJ·mol-1.结果表明: 水溶剂环境下,α-Ala的可以缓慢地旋光异构;在浓度较大的水汽环境下,α-Ala的旋光异构更容易进行.水汽环境下,羟自由基的存在可使α-Ala损伤;水溶剂环境下,羟自由基的存在可使α-Ala迅速损伤.
