微分电容法研究离子液体二氯乙烷混合体系的电化学双电层结构
2018-09-12赵金凤
赵金凤,周 尉
(上海大学 化学系,上海 200444)
室温离子液体是室温下完全由阴阳离子配对组成的液态盐,通常具有电化学窗口宽(可达5~6V)、热稳定性高、蒸汽压低等优点,在二次金属-空气电池、锂离子电池、电沉积和电容器等电化学器件和基础研究中引起了广泛的关注.然而,室温离子液体粘度高,电导率(0.1~18mS/cm)远小于传统水体系电解液,尤其季铵基、吡咯基、哌啶基和吡啶基离子液体的电导率更低,仅为0.1~5mS/cm.在离子液体中添加有机溶剂乙腈[1]、碳酸丙烯酯[2-3]等,是一种提高离子液体基混合体系电导率的有效途径,能够增强混合电解质体系的导电性能,同时很大程度上保留了离子液体本身的特性.1,2-二氯乙烷(DCE)作为一种常见的有机溶剂,其自身粘度较低(0.73cP),不易挥发(沸点83.5℃),而且具有较宽的电化学窗口,在有机电化学研究中被广泛应用.尤其,DCE与1-丁基-3-甲基咪唑三氟甲磺酸离子液体([Bmim][OTf])有较好的互溶性,是研究离子液体-有机溶剂混合体系的理想对象.
离子液体基混合电解液在电化学器件方面的应用离不开对其界面双电层(Electric Double Layer, EDL)结构的认识.目前,关于纯离子液体-金属电极界面结构的研究在理论和实验两方面都有报道.在理论研究方面,Kornyshev和Fedorov在平均场理论假设的基础上,忽略离子间的短程相互作用及离子液体体积在电场条件下的变化,建立了电极/离子液体界面电容的理论方程,讨论了离子液体中离子电荷传输和双电层区间的结构转变[4-5].Yan等拓展了平均场理论,提出了离子尺寸对离子液体中双电层结构和微分电容的影响,讨论了特性吸附的影响[6-7].在实验研究方面,研究者们用各种方法研究离子液体/电极的界面结构,包括微分电容法[8]、和频发生光谱[9](SFG)、现场红外光谱技术[10]、X射线反射[11]、原子力显微镜(AFM)[12]和扫描隧道显微镜(STM)[13]等.其中,利用电化学阻抗谱(EIS)测定电极界面的微分电容是一种经典且有效的电化学研究方法.Ohsaka等测量了离子液体[Pmim]BF4中汞电极、玻碳电极和金电极上的界面微分电容曲线[14],以及金(111)电极在离子液体[Emim]BF4、[Omim]BF4中的微分电容曲线,提出了在不同电位下的界面结构模型[15].Lockett等研究了不同碳链长度的离子液体[Cnmim][Tf2N]中金电极、铂电极和玻碳电极界面的微分电容曲线,讨论了温度、阴阳离子组成和电极材料对微分电容曲线的影响[16],以及电极电位的变化对阴阳离子在电极上吸脱附的影响和双电层结构的变化.Zhou等利用SFG研究了[Bmim][OTf]/铂电极的界面结构,并发现了离子液体体系所特有的阴阳离子吸脱附延迟效应[9].而类似的延迟效应在Gore等对[Bmim][OTf]/金电极的界面微分电容实验中也被观察到[17].
目前,对于离子液体混合体系的界面双电层的研究仅有一些初步的报道.Qiao等运用动力学模拟研究了离子液体[Bmim]BF4和乙腈混合体系的双电层结构,提出了“广义溶剂中的反电荷层”(CGS)的理论框架,来描述所研究的体系中的双电层结构[18].Bozym等测得[Emim][TFSI]分别与1,2-二氯乙烷、乙腈和碳酸丙烯酯混合后所得混合体系在玻碳电极上的界面微分电容曲线,通过比较微分电容曲线上的最小电容值,发现该电容值在离子液体中摩尔含量为5~10%时达到最大[19].由于纯离子液体和水溶液中的双电层模型并不适用于离子液体-有机溶剂混合体系,相关的电极界面结构的认识仍非常有限.
本文使用全频交流阻抗测试法研究了[Bmim][OTf]和DCE混合体系在多晶金电极表面的微分电容曲线.此前报道的纯离子液体体系的微分电容研究表明,单频交流阻抗测试对于所得到的微分电容曲线具有一定的不确定因素[20].由于多晶电极表面随频率改变会发生电容弥散现象,电极表面粗糙度、原子尺寸上的缺陷等因素增加了电流密度沿表面的不均匀分布,增加了电容的计算难度,导致单频数据计算存在不稳定性[21],因此本文采用了全频测试法.结果表明混合体系的电导率和微分电容值相比纯离子液体有明显的增加.同时发现在电化学窗口内随着电位的改变,阴离子[OTf]-在金电极表面发生特性吸附(Specific Adsorption, SA).本文根据所得的电化学窗口内的微分电容结果,讨论了相应的界面结构模型.
1 实验部分
1.1 仪器与试剂
CHI660A型电化学工作站(上海辰华仪器公司);Auto Lab PGSTAT30电化学工作站(瑞士万通公司);KQ2200B型超声清洗仪(昆山超声仪器有限公司),实验所用的离子液体[Bmim][OTf](99%)由兰州化学物理所购得,DCE(色谱级,99.9%)由阿拉丁购得,配制[Bmim][OTf]-DCE混合体系均在手套箱中进行.所用水均为超纯水.离子液体在使用前经过真空加热至110℃,以除去痕量水份.其他分析纯级别以上的试剂直接使用.
1.2 电化学部分
1.2.1 电极清洗
金圆盘电极(直径2mm)使用前用0.05μm氧化铝抛光粉打磨,并依次在超纯水、丙酮、乙醇和超纯水中超声洗涤,每次不超过2min,各重复3次后,将电极浸入浓硫酸中活化.实验前用超纯水清洗后在0.5mol/L硫酸溶液中进行电化学清洗,直至出现多晶金电极的标准循环伏安曲线.再用超纯水清洗,干燥后存于手套箱待用.
1.2.2 实验装置
电解池为自制的密闭玻璃池.金圆盘电极(直径2mm)为工作电极,自制铂片电极为辅助电极(1cm×6cm),铂丝电极为准参比电极,直接插入离子液体使用.辅助电极和铂丝电极采用和工作电极类似的电化学清洗过程进行处理.
1.2.3 电化学测试
循环伏安测试在CHI660A型电化学工作站上进行,交流阻抗测试是在Autolab电化学工作站上进行.为了保证在长时间的EIS测量过程中离子液体混合体系的稳定性,选择在双电层充放电特征明显的较窄的电位范围内进行测试.交流信号的幅度取10mV,频率范围为1~105Hz,在双电层电势区间内每隔50mV记录一次不同电位下的交流阻抗谱.为了使界面上的离子随电位变化有充足的时间达到重排再平衡,每个电位下EIS测定前的平衡时间设定为180s.本实验中,EIS测试是从负电位向正电位方向逐点进行.双电层电容(Cd)的计算是基于由电阻(R)和双电层电容(Cd)串联而成的界面等效电路.Cd值可由公式(1)推导出来
(1)
其中:f为阻抗测试频率;Z′是阻抗的虚部[22].得到全频测试结果后,通过对式(1)斜率的拟合来计算每个电位下的界面微分电容.
不同浓度溶液的电导率通过在开路电位(OCP)下的交流阻抗测试来获得[3].所有测试均在约20℃的室温下进行.使用20℃下电导率为0.0117S/cm标准KCl水溶液(0.1mol/L)测定电池常数.
2 结果与讨论
2.1 电导率测定

图1 [Bmim][OTf]-DCE混合体系电导率随离子液体浓度的变化Fig.1 Conductivities in mixtures of [Bmim][OTf] and DCE as a function of IL concentration
图1是[Bmim][OTf]和DCE混合体系中电导率随[Bmim][OTf]浓度变化的曲线.Zech等[23]测量了[Bmim][OTf]在不同温度下的电导率,其中在15℃时为1.8mS/cm,在25℃时为2.90mS/cm,在35℃时是4.36mS/cm.因此温度对电导率的影响很大,本实验测量的温度是在室温下(约20℃),测得的[Bmim][OTf](4.67mol/L)的电导率为2.7mS/cm,与上述文献报道相符.随[Bmim][OTf]摩尔浓度的降低,混合体系的电导率呈现先增加后减小的趋势,在2.5mol/L时达到了最大值(9mS/cm),是纯[Bmim][OTf]电导率的3倍多.电导率增加的原因可能是DCE的添加使得该混合体系比纯离子液体体系的粘度降低,导致电导率增加.随着离子液体浓度进一步降低到1mol/L以下,由于离子数目的减少,电导率明显下降.
2.2 循环伏安测定
图2为[Bmim][OTf]和DCE混合体系在金电极上的循环伏安图.实线部分为每个体系的电化学窗口.虚线部分为相应的EIS测试的电位区间,为保证在长时间的EIS测试中离子液体的稳定性,所选电位区间较窄.

图2 金电极在纯[Bmim][OTf]和[Bmim][OTf]-DCE 混合体系中的循环伏安图,虚线部分为电化学阻抗测试的电位区间Fig.2 CVs of neat [Bmim][OTf] and [Bmim][OTf]-DCE mixtures on Au electrode. The potential regions for EIS experiment are indicated by dotted lines
图2(a)为纯[Bmim][OTf]体系的电化学窗口,约4.4V(-2.5V~1.9V),和文献报道符合.当[Bmim][OTf]的浓度逐步降低至0.05mol/L时(图2(b)~(e)),[Bmim][OTf]和DCE混合体系的电化学窗口在阳极一端有负移的趋势,导致电化学窗口略有减小.值得注意的是,图2(d)、2(e)中,在-1.3V左右,出现一阴极峰,而图2(b)、2(c)中在该电位处未发现明显的单峰.将离子液体换成四丁基溴化铵盐(0.1mol/L)同样可观察到此峰.此阴极峰可能是随着DCE的浓度增加,DCE和[OTf]-在金电极上的竞争吸附逐渐处于优势而产生.曾有报道指出,DCE和在Pt电极表面上可发生吸附作用[24].因此,我们将-1.3V的阴极峰归属为金电极上DCE的吸附过程.
2.3 微分电容曲线测定
2.3.1 纯[Bmim][OTf]体系中的微分电容曲线

图3 金电极在纯离子液体[Bmim][OTf]中的界面微分电容曲线Fig.3 Differential capacitance curve of neat [Bmim][OTf] on Au electrode
图3为纯[Bmim][OTf]中金电极上的微分电容曲线.
在-0.1V和1.0V分别观察到两个单峰,根据Kornyshev的理论预测[5,25],对于阴阳离子尺寸相差不大且解离程度较大(γ>1/3)的离子液体,其微分电容曲线的形状为钟型,相应曲线中电容的最大值对应的电位归属为零电荷电位(PZC).据此,图3中电容最大值对应的电位(约-0.1V)可归属为零电荷电位.而在1.0V左右出现的电容峰很可能是因为在金电极表面大量吸附的[OTf]-阴离子所导致,[OTf]-中的SO3官能团在金电极表面发生特性吸附.如果只考虑双电层中的紧密层,根据Helmholtz模型,双电层微分电容的方程为
(2)
这里的ε和ε0分别为媒介中和真空中的介电常数,d是双电层厚度.显然,Cd值由d和ε决定.在25℃下,[Bmim][OTf]的ε为12.9±0.5[26],DCE的ε为10.37.而对于纯[Bmim][OTf]体系,Cd由d值大小而变化.由[OTf]-特性吸附所形成的排列有序致密的紧密层具有较小的双电层厚度,从而导致电容值的迅速上升,形成了1.0V附近的电容峰.
另一方面,图3中,较PZC更负的电位区域的电容值显著降低,大大低于[OTf]-阴离子发生特性吸附的正电位区间.这个差异主要由于阴阳离子尺寸差异以及在电极表面的吸附能力造成.从电化学窗口的阴极端开始(-2.3V),电极表面的第1层吸附的是离子尺寸较大、且吸附能力较弱的[Bmim]+.而且由于较大的离子尺寸,为达到静电平衡,很可能在第2层中还会存在[Bmim]+,和[OTf]-的共同形成界面上的第2层离子排布,从而形成了相对较厚的双电层以及较低的微分电容值.当电位逐渐正向移动到较PZC更正的区域时,带有长烷基链的[Bmim]+开始从金电极表面脱附,[OTf]-逐渐取代[Bmim]+吸附在电极表面上,导致1.0V附近[OTf]-特性吸附峰的产生.
2.3.2 [Bmim][OTf]-DCE混合体系的微分电容曲线
图4是[Bmim][OTf]-DCE混合体系中金电极上的微分电容曲线.与纯[Bmim][OTf]体系相类似的是,[Bmim][OTf]-DCE混合体系的微分电容曲线也呈现出钟形特征.在-0.2V~0V范围内出现的较大的电容峰类似地归属于零电荷电位PZC.与纯离子液体中的PZC电位相比,混合体系中的PZC发生负移(图4(a~(c)).在纯离子液体体系中,开路电位下金电极表面易被[Bmim]+吸附.然而在混合体系中,DCE的存在使得咪唑环上的π电子与金电极上的空轨道重叠度减小,从而[Bmim]+与金电极的吸附作用减弱,导致PZC相比纯离子液体发生负移.
其次,混合体系的微分电容曲线另一重要特征是随着[Bmim][OTf]浓度的降低,1.0V附近的特性吸附峰也发生明显的减弱,说明随着[OTf]-阴离子数量的减少,金电极表面上由[OTf]-的吸附生成的紧密层结构发生变化,不再能形成类似纯离子液体中的致密的紧密层结构,从而导致正电位区域的双电层电容值下降.
再次,相比于纯离子液体,较高浓度的混合体系电容值在PZC附近发生明显增加,在1mol/L时达到了最大值(约110μF/cm2).为了研究离子液体中添加有机溶剂对电容的影响,Bozym等提出了初步的理论模型,认为溶剂引入后使得界面处电荷更加聚集,电容增加[19].Li等观察到双阳离子离子液体-乙腈混合电解液中发生类似的界面电容增加现象,并利用分子动力学进行了模拟计算,结果表明电容值的增加是由于引入乙腈后,同离子,即与电极所带电荷电性相同的离子,在双电层中发生较强的逐出作用,从而导致双电层的紧密层更加致密和电容值的上升[27].本文中,在[Bmim][OTf]-DCE混合体系中,DCE的引入促进了阴阳离子对的分离,导致同离子从双电层中的逐出,从而反离子更加容易吸附在电极表面.其结果导致双电层厚度减小和电容值的增加.然而,当[Bmim][OTf]浓度降至0.05mol/L时(图4(d)),PZC处的电容峰值减少至约30μF/cm2,这可能是由于离子数目的减少和离子排列不够紧凑.在较低的浓度下,紧密层和分散层的界限不明显,紧密层也很可能消失,导致整体的双电层厚度增加,混合后的介电常数减小,根据方程(2),微分电容值减小.其次在1mol/L(图4(b))时,-1.2V有个较小的电容峰,这可能是由于在DCE的作用下,[Bmim]+在该电位下与金电极产生了相互作用导致双电层结构变化.
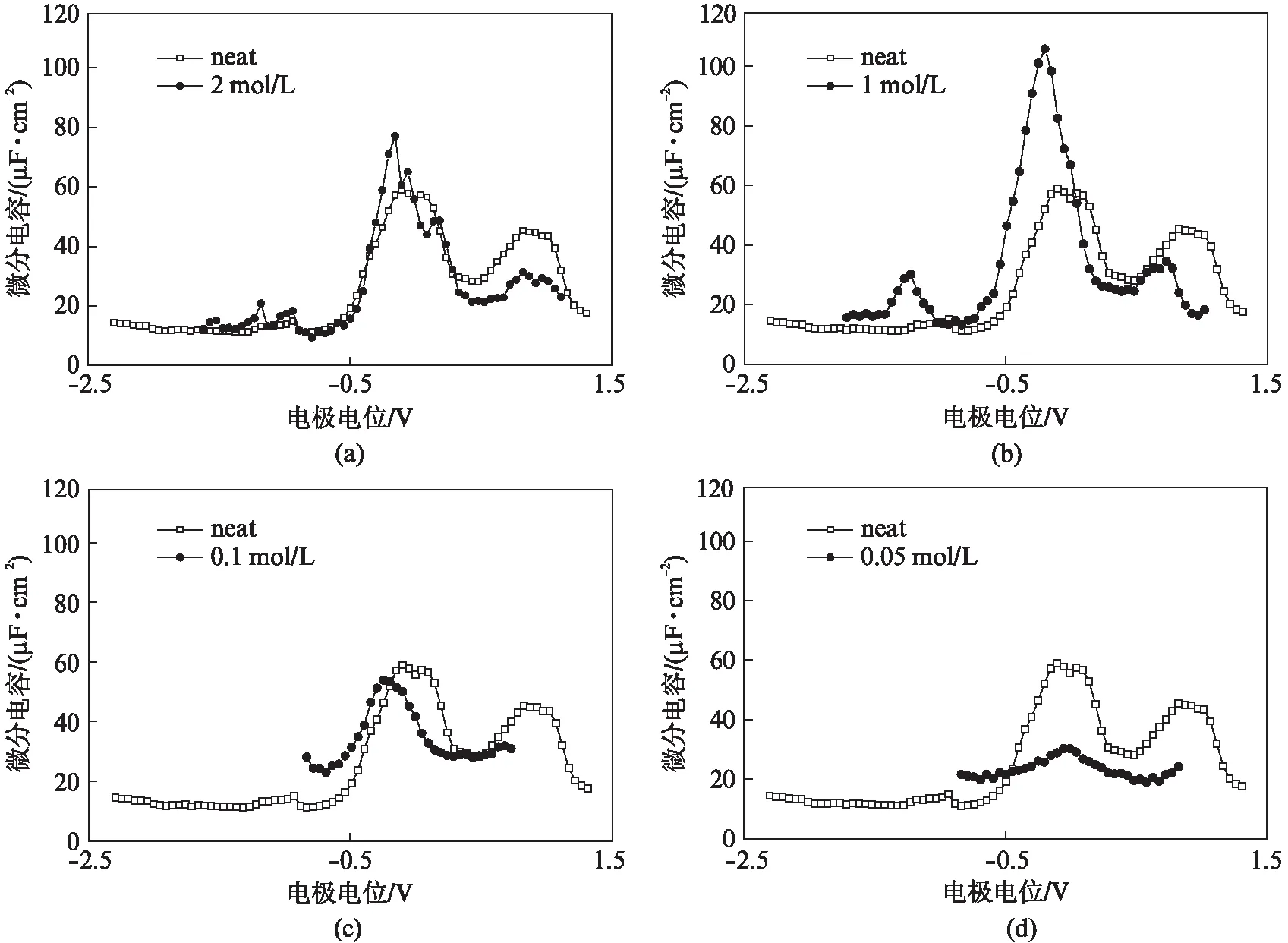
图4 金电极在不同浓度的[Bmim][OTf]-DCE混合体系中的微分电容曲线Fig.4 Differential capacitance curves on Au electrode in mixtures of [Bmim][OTf]-DCE of different concentration
2.3.3 界面结构模型的讨论
目前为止,微分电容测定和计算模拟等多种研究方法证明由离子液体形成的电极界面结构倾向于由多层离子形成的双电层结构[28].在这里,我们认为[Bmim][OTf]和DCE混合体系形成的电极-电解液界面的双电层为多层结构,厚度依赖于离子的结构和尺寸.图5(见第514页)给出了纯离子液体体系和混合体系中双电层结构模型.图5(a)和5(b)是纯[Bmim][OTf]中双电层结构模型.在负电位下,第1层中[Bmim]+优先吸附在电极表面,第2层吸附的是[OTf]-和小部分[Bmim]+,而[Bmim]+体积较大,造成了较厚的双电层.相反地,在正电位下,电极表面的第1层是[OTf]-,第2层大多是[Bmim]+,由于[OTf]-尺寸小,双电层厚度较小.在DCE存在时,混合体系的电容值比纯体系的电容值大.图5(c)和5(d)是[Bmim][OTf]-DCE混合体系中双电层结构模型,5(c)是低浓度下的界面结构模型图,5(d)是高浓度下的界面结构模型图.[Bmim][OTf]的介电常数比DCE的介电常数大,二者以不同的比例混合时,介电常数值也会改变.相比于[Bmim][OTf]的介电常数,当离子液体浓度较高时,混合体系的介电常数会略有降低,此时的微分电容主要受双电层厚度的影响.当离子液体浓度较低时,混合体系的介电常数会明显降低,此时的微分电容主要受介电常数和双电层厚度的影响.
在正电位下,当[Bmim][OTf]浓度较低时,离子数目的减少使得双电层中离子的排列不紧凑,紧密层可能消失,仅有分散层存在,双电层较厚,介电常数降低,根据方程(2),双电层电容会明显减小.[Bmim][OTf]浓度较高时,[OTf]-有序地排列在电极表面,第2层DCE和[Bmim]+交错排列,双电层较薄,造成微分电容增加.造成这种排列方式的可能原因是DCE具有逐出同离子的能力,从而促进反离子排列在电极表面.
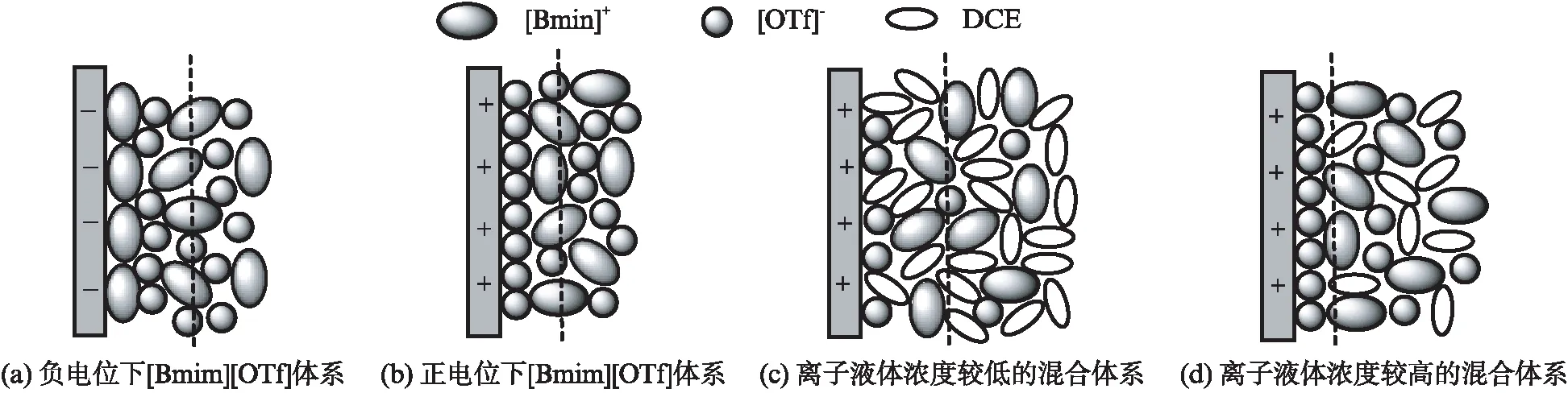
图5 纯[Bmim][OTf]体系和[Bmim][OTf]-DCE混合体系的双电层结构示意图Fig.5 Schematic diagrams of EDL structures in neat [Bmim][OTf] and mixtures of [Bmim][OTf]-DCE
3 结 论
本文进行了纯[Bmim][OTf]体系和[Bmim][OTf]-DCE混合体系中金电极上的电化学研究.电导率测试结果表明,随着[Bmim][OTf]摩尔浓度的降低,溶液电导率先增大后减小,在2.5mol/L时达到最大,这说明添加低粘度的DCE可以有效的改善离子液体基混合电解液的导电性.循环伏安测试结果表明,随着[Bmim][OTf]摩尔浓度的降低,电化学窗口并没有明显的变窄.EIS测试结果表明,DCE的存在下,混合体系电容值明显增加,在1mol/L时达到了最大.随电位的改变,在较正电位下,[OTf]-在金电极表面有特性吸附导致界面结构急剧变化.为了解释这一现象,我们提出了相应的界面双电层结构模型.
