利用epiCRISPR载体表达双sgRNA实现高效的HDAC基因编辑
2018-09-12王子实李苗苗王永明
王子实,李苗苗,王永明
(复旦大学 生命科学学院,上海 200438)
基因编辑技术(genome editing)是在基因组水平上对DNA进行修饰的遗传操作技术.目前最常用的基因组编辑技术是CRISPR(Clustered regularly interspaced short palindromic repeats)/Cas9技术[1].CRISPR/Cas9系统包括2个元件,分别是具有核酸内切酶活性的Cas9蛋白和向导RNA(guide RNA, gRNA).gRNA是将crRNA和tracrRNA融合得到的,其中crRNA负责识别靶序列,tracrRNA负责结合Cas9蛋白[2].当gRNA和Cas9蛋白结合后,gRNA序列就会将Cas9蛋白引导至和其序列互补的基因组区域,使得Cas9蛋白和目标基因组序列结合,并切断基因组DNA产生双链断裂(double-strand break, DSB)[3].细胞内修复DNA双链断裂的2种主要途径分别是非同源末端连接(nonhomologous end joining, NHEJ)和同源重组(homologous recombination, HDR)[4].前者经常引起碱基的缺失或者插入(indel),后者会精确地改造基因组序列[5-6].使用CRISPR/Cas9技术进行基因敲除主要是通过NHEJ途径实现的,NHEJ引发的indel会造成目标基因发生移码突变,其概率为2/3.纯合敲除细胞系的构建要求目标基因的所有拷贝都发生移码突变.理论上对于双倍体细胞基因组,2个等位基因都发生indel引发的移码突变的概率是(2/3)2.而如果目标基因在基因组中存在多个拷贝,则编辑后构建纯合敲除细胞系的概率就进一步降低为(2/3)n(n是基因拷贝数).所以拷贝数越多,敲除难度越大.
传统的基因编辑技术使用普通的质粒表达Cas9和gRNA,是瞬时表达系统.质粒随着细胞的传代扩增而被稀释,使得Cas9和gRNA在细胞内的表达水平下降,进而降低了基因编辑的效率.为了克服上述缺点,本实验室使用附着体载体(episomal vector)表达Cas9和gRNA,称为epiCRISPR系统[7].附着体载体可以在真核细胞中扩增[8],能够长期表达外源基因.epiCRISPR除了表达Cas9和gRNA,还表达嘌呤霉素抗性基因,在药物的筛选下,能够富集转染成功的细胞,提高获得纯合敲除细胞系的效率.同时附着体载体不整合到基因组中,编辑完成后,撤除筛选药物,质粒会很快丢失,细胞不再表达外源基因.利用epiCRISPR技术,可以实现平均80%以上的indel效率.
利用CRISPR/Cas9编辑基因时,编辑不一定会造成基因功能丧失.尽管基因编辑可能会导致多种形式的DNA序列改变,包括碱基置换和indel.但碱基置换可能并不会造成氨基酸的改变;即使氨基酸发生了改变,如果不是重要功能域的氨基酸,也不会使基因功能丧失.indel一般在几十个碱基以内,如果长度是3的倍数,会造成氨基酸数量的改变,但不一定会造成移码突变.敲除基因的有效方法是在基因编码区前端进行编辑,当indel长度不是3的倍数时,会造成移码突变,蛋白的翻译提前终止,进而导致功能丧失.如果在基因上设计2个gRNA,它们中间删除的长度不是3的倍数,会增加移码突变的概率[9].设计2个gRNA还有另外一个优点,不同gRNA的活性差异很大,2个gRNA中的一个活性高就可以敲除基因,如果2个活性都高,敲除效率会更高.
HDAC全称是组蛋白去乙酰化酶(histone deacetylase),其主要功能是对染色质中的组蛋白去乙酰化,与DNA复制、修复以及染色质结构都有密切的关联[10].HDAC在生物体内和很多基因的表观遗传状态有关[11],是阿尔茨海默症等神经退行性疾病[12]以及黑色素瘤[13],乳腺癌[14]等多种癌症的潜在治疗位点[15].HDAC家族包含11个基因,分别在不同的代谢过程中负责组蛋白的去乙酰化,是一个作用广泛、功能多样的基因家族.因此,构建HDAC纯合敲除细胞系对于研究HDAC基因家族的功能是十分必要的.在本研究中,我们通过敲除HDAC家族基因探讨附着体载体表达双gRNA系统的有效性.一方面可以快速构建HDAC基因家族纯合敲除细胞系,为将来研究HDAC家族基因功能铺垫良好的基础.另一方面,本实验还可以从技术层面对结合使用epiCRISPR技术和双gRNA技术的基因敲除效果进行验证.
1 材料和方法
1.1 材料
本文中使用的细胞系为神经胶质瘤母细胞癌细胞系SK-N-BE(2)和HeLa细胞.转化用的大肠杆菌E.coliDH5α来源于TIANGEN.所有实验用到的限制性内切酶均来源于NEB.PCR引物合成以及DNA测序由Genewiz苏州分公司完成.Western blot使用抗体来自Abcam,Santa-Cruz以及CST.
1.2 方法
1.2.1 细胞培养
神经胶质瘤母细胞癌细胞系SK-N-BE(2),培养基使用MEM(Gibco)+10% F12(Gibco)+10% FBS(Gibco),培养条件37℃,5% CO2.HeLa细胞系,培养基使用DMEM(Gibco)+10% FBS(Gibco),培养条件37℃,5% CO2.2种细胞均使用Thermo Nunclon系列一次性细胞培养皿进行培养.
1.2.2 CRISPR/Cas9系统gRNA以及基因编辑检测PCR引物的设计
质粒构建过程中使用的所有内切酶以及连接酶均来源于NEB.使用http:∥crispr.mit.edu: 8079/?网址提供的gRNA设计工具,在目标基因的外显子区域设计临近的2段gRNA作为实验使用的gRNA.并且在gRNA目标区域附近设计PCR引物,用以在PCR后使用T7E1(NEB)酶切检测编辑效率.表1(见第414页)展示了实验中使用的gRNA以及相应的PCR连接引物以及检测引物.引物名称中数字代表gRNA间距.
1.2.3 转染试剂与细胞转染
转染试剂: 转染试剂使用Lipofectamine©2000(Invitrogen).
转染流程: 细胞铺板,当细胞覆盖培养皿底部60%~70%时进行转染.对于常见的6孔板培养的细胞转染而言: 样品1,使用150mL Opti-MEM(Gibco)与5μL Lipo 2000(Invitrogen)充分混合.样品2: 150mL Opti-MEM(Gibco)与2000ng待转染质粒充分混合,样品1,样品2在各自室温静置5min后充分混合到一起,再次室温静置20min.之后将混合样品均匀滴加至细胞培养皿内部.轻微摇晃培养皿使转染试剂与培养基混匀.转染17~20h后,更换新鲜的培养基进行培养,并使用相应的药物对细胞进行筛选.
1.2.4 单克隆细胞分选与培养
开始转染为第0天.在使用药物筛选10天后,使用细胞计数法对编辑后的细胞进行稀释分选,确保96孔板(Thermo)平均每孔中有一个细胞.在第15天观察每孔中实际细胞数量和细胞存活情况,确认真正的单克隆数量.第20天后将生长正常的单克隆进行传代,能够存活到2次传代以上的单克隆可以认为是一个稳定的单克隆.

表1 HDAC基因家族纯合敲除细胞建立过程中使用的gRNA以及PCR引物
1.2.5 单克隆细胞编辑效果测序鉴定
根据基因组上的序列,在目标基因的编辑位点附近根据巢式PCR原则设计检测PCR引物.并使用Genome Quick Extraction(Epicentre)试剂回收第20天单克隆细胞基因组.使用设计好的PCR引物对提取的单克隆基因组进行PCR,得到编辑后的目标基因序列(500~700bp长度)的PCR产物.再通过的pGEM-T Easy T(ProMega)载体连接系统将PCR产物连接到T载体上.使用TIANGEN产E.coliDH5α将连接产物进行转化扩增,并通过T载体测序结果确认单克隆的编辑情况.测序结果使用Vector NTI软件进行序列比对.
1.2.6 Western blot
使用SDS-PAGE检测基因敲除前后HDAC基因家族的蛋白表达水平.电泳完成后使用转膜仪(BioRad)进行转膜.之后使用封闭液(1×TBS+0.5% Tween)进行封闭,封闭完成后加入一抗(表2),再加入二抗进行杂交(表2),最后使用显色液(Tanon)进行显色.

表2 HDAC基因家族蛋白分子量及Western blot中使用的抗体
2 结果与分析
2.1 含有双gRNA的epiCRISPR质粒的设计与构建
本实验中使用的CRISPR-Cas9质粒是自行设计的epiCRISPR质粒,其基本结构如图1所示.其中,hU6启动子起始gRNA的转录;EFS启动子起始Cas9、嘌呤霉素抗性基因(puro)和绿色荧光蛋白基因(GFP)的转录,这3个基因位于同一个阅读框内,由2A片段隔开.EBNA1是基因组黏附元件,可以将质粒黏附于基因组上,使得质粒伴随着基因组的复制而复制,提高质粒的表达效率[8].
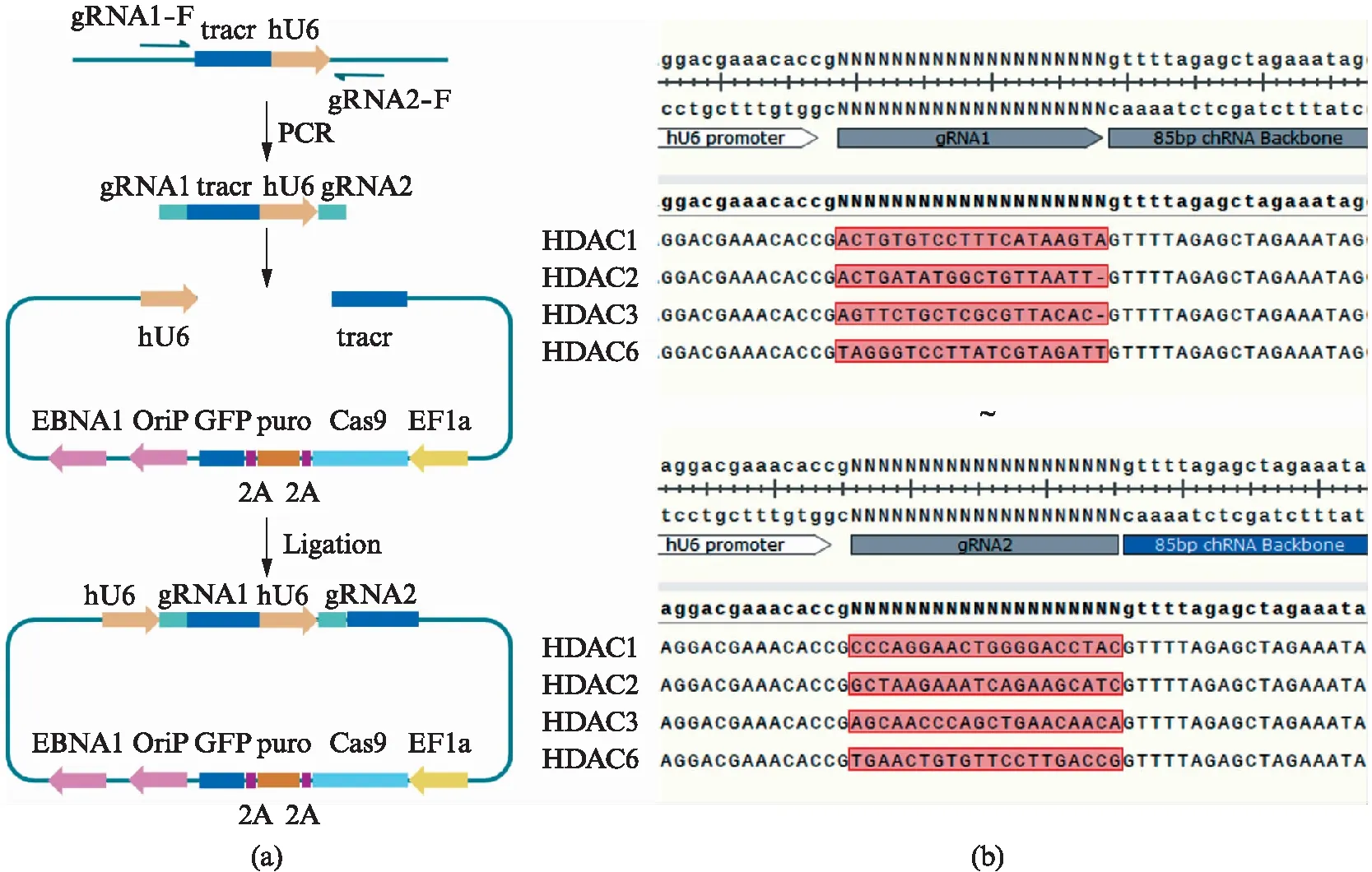
图1 含有双gRNA的epiCRISPR系统Fig.1 The epiCRISPR system contains two gRNAs(a) 含有双gRNA的CRISPR系统的构建;(b) 双gRNA插入测序确认.
我们发明了在一个epiCRISPR质粒上连接2个gRNA的方法.质粒上有2个BspQ1酶切位点,位于hU6和tracrRNA之间.如图1所示,在这个地方插入crRNA1-tracr-hU6-crRNA2序列,就可以表达2个完整的gRNA.为了实现这个目的,我们设计了一个中间载体,含有tracr-hU6序列.再设计一对与tracr-hU6序列匹配的引物,分别含有crRNA1和crRNA2,同时都含有BspQ1酶切位点.经过PCR扩增就得到了crRNA1-tracr-hU6-crRNA2序列,酶切后就可以连接到epiCRISPR质粒上了.
为了进一步提高epiCRISPR质粒系统的基因敲除效率,我们对原有的epiCRISPR系统进行了改造.目的是在epiCRISPR质粒上同时表达2个gRNA,进一步提高系统的基因编辑能力.由于Cas9蛋白本身没有使CRISPR序列成熟的核酸酶功能,必需借助RNAseⅢ的帮助才能切割串联的CRISPR序列(等效于串联的gRNA+tracr序列),使串联的CRISPR成熟,形成可以和Cas9蛋白结合的gRNA序列[3].所以不能简单地通过前后串联2个gRNA来实现不同的gRNA表达.对于外源的CRISPR/Cas9系统而言,为了形成不同的gRNA,必须使用不同的外源启动子单独起始表达不同的gRNA.因此我们在原有gRNA插入位置插入了gRNA1+hU6启动子+gRNA2的片段,形成了2个hU6启动子分别负责gRNA1和gRNA2的转录.这样我们在一个质粒上就实现了2种gRNA的同时表达(图1).
我们设计的gRNA有如下特点: (1) 2个gRNA之间的碱基数量不是3的倍数(表1),如果中间的部分被切掉后,准确地连接在一起能够形成移码突变;(2) 2个gRNA距离很近,都在一个外显子上,用一对引物就可以检测编辑的结果.
为了证明epiCRISPR系统结合双gRNA确实能够增加基因编辑的效率,我们设计了一组对照实验.分别使用含有EBNA1元件和双gRNA的质粒1以及含有同样双gRNA但是不含有EBNA1元件的质粒2(图2(a))同时转染HeLa细胞,按照1.2节中步骤分别取转染后第5天和第10天样品进行编辑效率检测.检测结果如图2(b)所示.可以看出在第5天的时候,质粒1和质粒2的编辑效率没有明显区别(lane 2和4).但是经过药物筛选后的第10天,含有EBNA1的质粒1由于质粒本身能够随着细胞基因组复制,因此分裂后的细胞依然继承了抗性,因此随着筛选时间的增加,编辑效率也随之增加(lane 3).而不含有EBNA1元件的质粒2转染的细胞由于质粒丢失,细胞会逐渐被抗性药物杀死(图2(c)).虽然第10天的编辑效率才59%(图2(b)lane 3),但是事实上,由于我们的系统使用的是双gRNA,根据之前的文献[9]研究结果,使用临近的双gRNA对目标基因组进行编辑的时候,有较高的概率会恰好将2个gRNA中间的片段完全去除,也就是说使用临近的双gRNA编辑的结果中有很多完全相同的编辑,这些相同的结果会被T7检测误判为未编辑.也就是说第10天的编辑结果要远大于59%.

图2 EBNA1元件增强基因编辑效率Fig.2 EBNA1 enhances gene editing efficiency(a) 质粒1和质粒2结构示意图;(b) 基因编辑效率检测.1. DNA ladder 1000;2. 质粒1第5天;3. 质粒1第10天;4. 质粒2第5天;5. 阴性对照,野生型基因编辑效率检测;(c) 抗性筛选第10天细胞拍照,不含有EBNA1元件的细胞由于质粒丢失已经完全被抗性药物杀死(bar=500μm).
2.2 HDAC1纯合敲除细胞系的构建
HDAC1作为HDAC基因家族的成员之一,在真核生物基因表达的调控中起重要作用.HDAC1蛋白是组蛋白乙酰化复合体的成分之一[16].并且,HDAC1能够增强肺上皮细胞对于A型流感病毒的抵抗[17].HDAC1和肺癌的发生发展存在重要的关联[18],HDAC1的敲除对于食道癌的治疗也有积极的意义[19].因此,我们尝试使用结合了双gRNA设计的epiCRISPR系统建立HDAC1纯合敲除细胞系.
根据图3(a)所示流程,我们将质粒转染到了神经胶质瘤母细胞癌细胞系SK-N-BE(2)中.转染20h后加入puromycin进行筛选,筛选后第10天进行单克隆分选,之后培养至第20天,选择能够稳定传代的细胞系进行测序和Western blot分析.
从实验结果(图3(c),(d))中可以看出,使用含有双gRNA的epiCRISPR系统敲除HDAC1基因,在得到的7个单克隆中,有3个是纯合敲除的单克隆(Clone 5,6,7).测序结果为野生型的Clone1,2,4在对应的Western blot泳道中存在明显的HDAC1蛋白条带.测序结果为纯合敲除的Clone 5和6在对应泳道中没有HDAC1蛋白的条带.而另一个测序结果为纯合敲除的Clone 7则表现为部分敲除,可能是因为Clone 7的敲除并不完全,HDAC1仍然有部分表达(图3).纯合敲除的细胞生长极其缓慢,说明这个基因对细胞生长很重要.一部分克隆在培养中死掉,推测也是纯合敲除造成的结果.
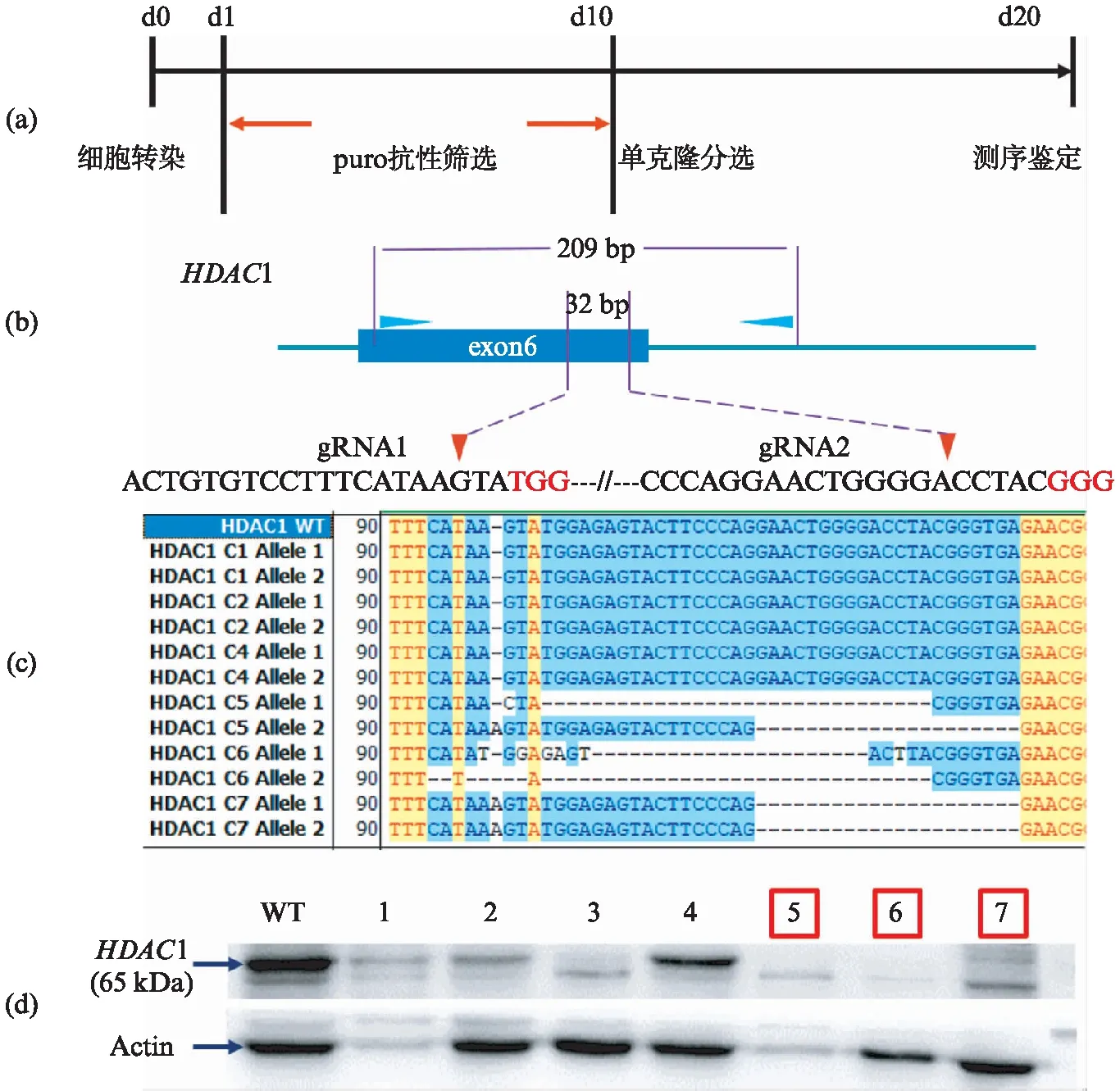
图3 HDAC1纯合敲除细胞系的建立Fig.3 The screening of HDAC1 homozygous knockout(a) HDAC基因家族纯合敲除细胞系筛选流程示意图;(b) HDAC1双gRNA设计以及检测引物位置;(c) 单克隆测序结果展示;(d) 单克隆Western blot检测.红色框标明纯合敲除,蓝色框标是杂合敲除.
2.3 HDAC2纯合敲除细胞系的构建
与HDAC1类似,HDAC2是组蛋白乙酰化复合体的另一个主要成分[16].HDAC2对很多癌症的耐药性都具有重要的影响,如对卵巢癌[20]和结肠癌[21]对化疗药物的敏感性都具有重要作用.
和HDAC1一样,按照图3(a)的流程进行单克隆筛选,测序之后进行Western blot验证(图4,见第418页).实验结果表明,和HDAC1类似,使用含有双gRNA的epiCRISPR系统进行HDAC2纯合敲除,筛选的效率也比较高,10个单克隆中得到了2个纯合敲除细胞系(Clone 8,10),这2个细胞系对应的Western blot结果没有HDAC2蛋白条带,说明这2个单克隆做到了HDAC2基因的完全敲除.另外还得到了2个杂合敲除细胞系(Clone 7,9).相对于野生型细胞系,杂合敲除的细胞系中HDAC2蛋白表达量存在明显的下降(图4(d)).纯合敲除的细胞也是生长极其缓慢.
2.4 HDAC3纯合敲除细胞系的构建
和HDAC1与HDAC2不同,HDAC3并不是组蛋白乙酰化复合体的主要组成部分,HDAC3主要在各种炎症因子相关的生物反应中起作用.HDAC3在风湿性关节炎相关的炎症反应中调节炎症基因的表达[22].HDAC3在吸烟引发的胰腺癌中也具有诱导和促进胰腺癌细胞增殖的作用[23].同时HDAC3也是一些发育过程的必要基因,例如,HDAC3是造血干细胞前体细胞DNA复制的必要调节基因[24].
但是按照图3(a)流程,我们无法得到一个稳定传代的HDAC3纯合敲除SK-N-BE(2)细胞系(图5,见第418页).存活单克隆的测序结果全部为野生型.之后,为了验证这个实验结果,我们使用HeLa细胞按照图3(a)流程进行同样的HDAC3基因敲除实验,也无法得一个HDAC3纯合敲除HeLa细胞系(数据未展示).考虑到HDAC3在生物体内以调控炎症相关基因并调节一些发育过程中重要前体细胞的增殖[24],并且有文献显示使用Cre重组酶条件敲除HDAC3会导致细胞分裂S期异常以及DNA损伤修复异常,进而导致细胞死亡[25].而使用类似技术在小鼠神经系统中敲除HDAC3的结果显示绝大部分小鼠在出生后24h内死亡[26].基于以上结果,我们认为HDAC3是一个纯合敲除致死基因,或者严格地说,至少对于SK-N-BE(2)细胞和HeLa细胞而言,HDAC3是一个纯合敲除致死基因.
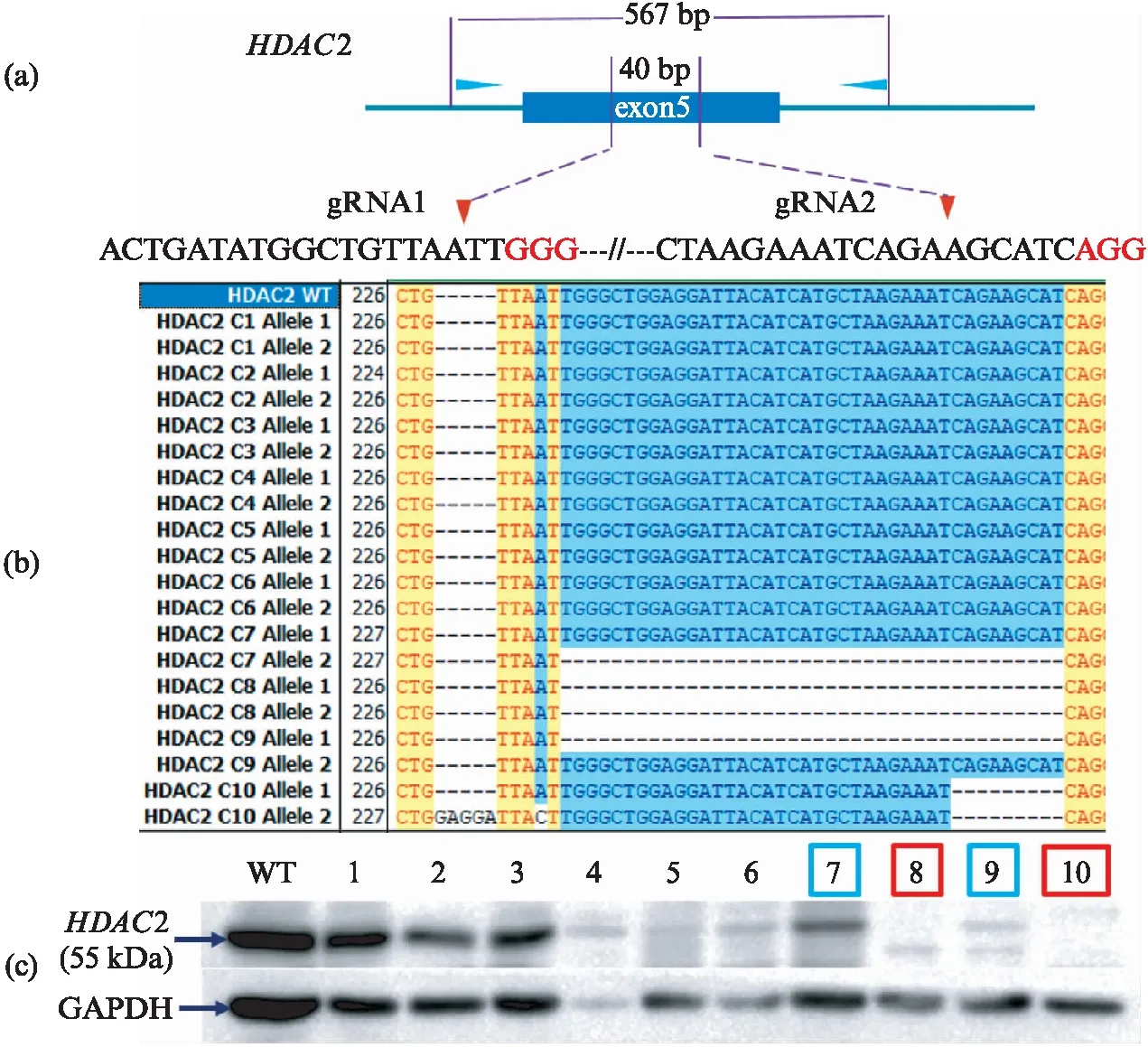
图4 HDAC2纯合敲除细胞系的建立Fig.4 The screening of HDAC2 homozygous knockout(a) HDAC2双gRNA设计以及检测引物位置;(b) 单克隆测序结果展示;(c) 单克隆Western blot检测.红色框标明是纯合敲除,蓝色框标明是杂合敲除.
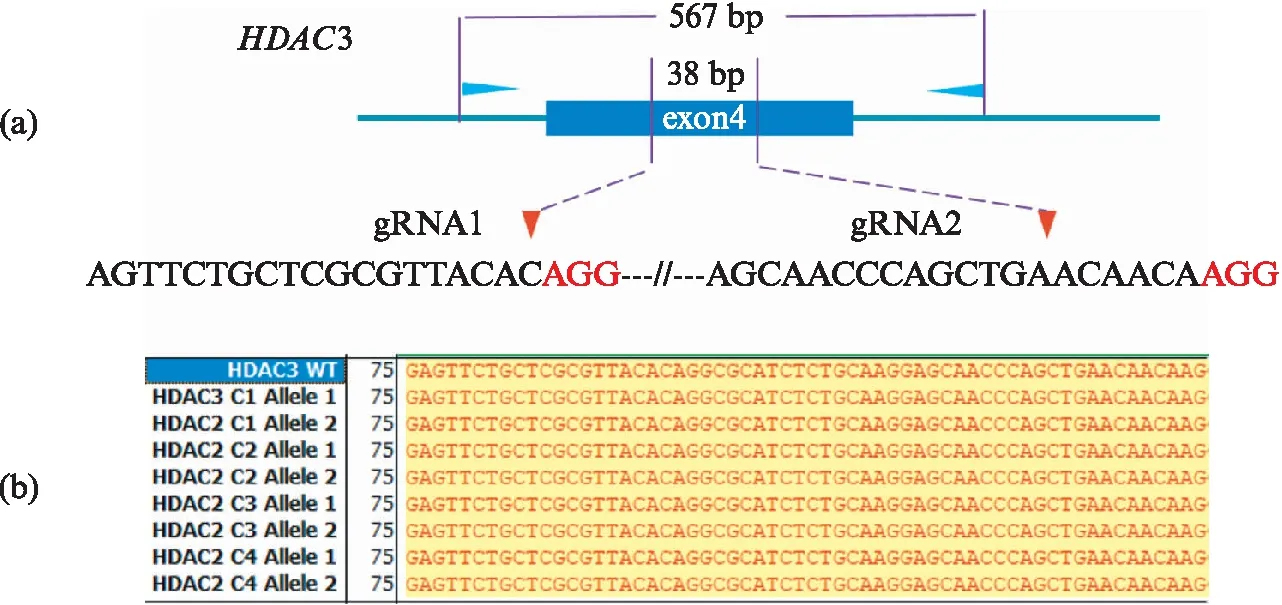
图5 HDAC3纯合敲除细胞系的建立Fig.5 The screening of HDAC3 homozygous knockout(a) HDAC3双gRNA设计以及检测引物位置;(b) 纯合敲除单克隆测序结果举例.
2.5 HDAC6纯合敲除细胞系的构建
和HDAC1,HDAC2不同,HDAC6也不是组蛋白乙酰化复合体的组成部分,它主要在细胞防御与病毒介导方面起作用[27].HDAC6的过量表达往往会促进癌细胞的增殖,并增强癌细胞对于化疗药物的抵抗,这种作用在恶性胶质瘤[28]和肺腺体癌[29]中都有过相关报道.
和HDAC1类似,我们同样按照图3(a)的流程对HDAC6基因进行纯合敲除单克隆筛选,测序之后也进行Western blot验证(图6).实验结果表明,使用含有双gRNA的epiCRISPR系统进行HDAC6纯合敲除,在一轮筛选中得到的5个单克隆中,有2个是纯合敲除细胞单克隆.这依然是一个非常高的比例.
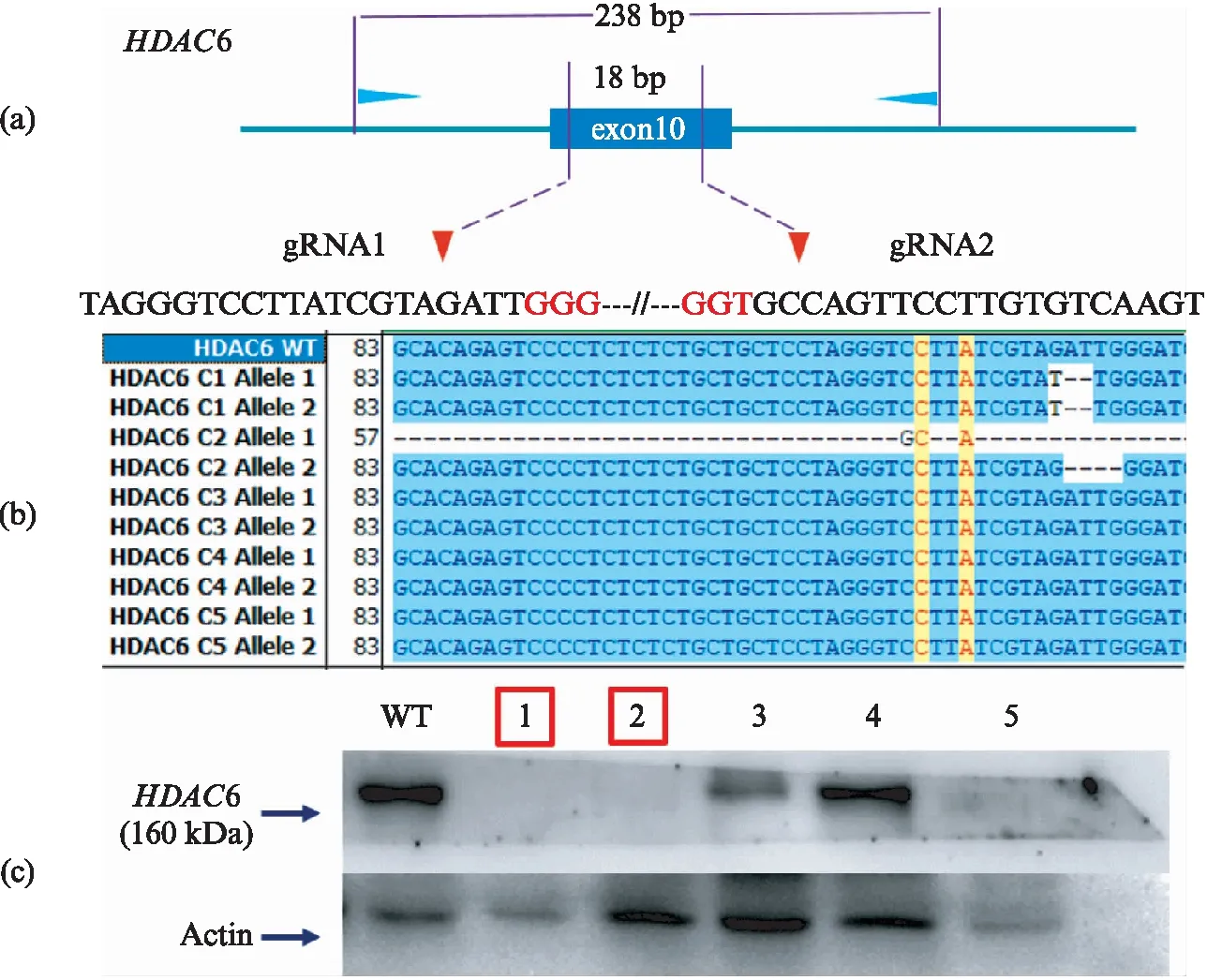
图6 HDAC6纯合敲除细胞系的建立Fig.6 The screening of HDAC6 homozygous knockout(a) HDAC6双gRNA设计以及检测引物位置;(b) 单克隆测序结果展示;(c) 单克隆Western blot检测.红色框标明是纯合敲除,蓝色框标明的是杂合敲除.
3 讨 论
本实验利用epiCRISPR表达双gRNA的技术,在SK-N-BE(2)细胞系中对HDAC基因家族进行敲除,目的是高效的得到HDAC基因家族纯合敲除细胞系.有报道表明,如果2个gRNA的位置毗邻的话,则有较大的概率会使得DNA在断裂处发生平末端连接而不是indel[30],导致2个gRNA之间的片段丢失,造成移码突变.并且由于2个临近的gRNA往往设计在同一个外显子上,即使没有片段丢失,2个gRNA造成阅读框发生错位突变的概率也远大于单独一个gRNA.
HDAC基因家族作为表观遗传学中重要的调节基因,通过对组蛋白乙酰化水平的调节影响多种基因的表达,进而参与多种细胞功能,如细胞增殖,细胞凋亡等,这使得HDAC基因家族成为多种疾病的潜在治疗靶点.因此,快速有效的建立HDAC基因家族纯合敲除细胞系,是进一步研究HDAC基因家族的生物学功能的必要基础.
最终的实验结果中,HDAC1,HDAC2和HDAC6都得到了符合预期的纯合敲除细胞系,并且纯合敲除的比例高达40%.这个比例和单独的epiCRISPR实验[8]以及双gRNA敲除实验[9]相比,有了明显的提高,说明epiCRISPR和双gRNA敲除这两种技术的结合可以有效地提高获得纯合敲除单克隆的概率.
值得指出的是,我们目前采取的流程是先得到一个稳定遗传的细胞系再进行测序确认.在实际的操作中,我们发现很多单克隆在生长和传代的过程中逐渐死亡.这部分死亡的单克隆并没有在统计结果中体现.推测这一中途死亡的单克隆里面有较大部分是纯合子,而杂合子和野生型的单克隆并不容易死亡.这就导致了我们目前的统计结果中,纯合子的比例要低于实际编辑结果.因此,如果不考虑HDAC3,目前使用带有双gRNA的epiCRISPR质粒得到的单克隆中,有接近40%的比例是纯合敲除.
与家族中其他基因不同,实验中HDAC3依然没有得到稳定遗传的细胞系.虽然有部分HDAC3纯合敲除细胞系在进行单克隆分选的时候能够存活,但是无一例外的,这些细胞系都没有办法稳定传代,形成可用的单克隆.因此,我们认为,至少对于SK-N-BE(2)细胞系和HeLa细胞系而言,HDAC3是一个纯合致死基因.
在本研究中,我对HDAC家族基因进行了敲除实验,高效快速地得到了HDAC家族基因的纯合敲除细胞系.考虑到CRISPR系统本身的广泛应用性,这种将epiCRISPR系统和双gRNA技术结合起来的方法也能够适用于其他基因的敲除,可以用于快速构建多种基因的纯合敲除细胞系,极大地扩展了本实验技术的应用范围.
